
Abstract
Synovial sarcoma (SS) is a soft tissue, generally deep seated neoplasms that occurs generally in the proximity of large joints. We report of a case of a 33-year-old man who was diagnosed with primary SS of the kidney which is an extremely rare tumor that accounts for less than 2% of malignant renal tumors. Contemporary management of renal synovial sarcoma includes surgical resection and ifosfamide-based chemotherapy and they remain the mainstay of therapy of synovial sarcoma, which is often applied, combined as part of an aggressive treatment approach. Fewer than 50 patients have been described in the English literature. Physicians should be aware of the possibility of malignancy in cystic renal masses and raise the suspicion of synovial sarcoma, especially when patients with renal masses are young adults. Along with the case report a literature review on primary synovial sarcomas of the kidney is provided with focus on the renal tumors’ differential diagnosis.
Introduction
Synovial sarcomas (SSs) account for 5-10% of adult soft tissue sarcomas and occur mostly in the proximity of large joints.1,2 SSs have been reported in other unusual sites including the thoracic and abdominal wall, head and neck region, retroperitoneum, bone, lung, or prostate.3,4 Primary SS of the kidney is an extremely rare tumor, first described by Faria et al. in 1999 and published by Argani et al. 5 Histopathological diagnosis is difficult, and always requires immunohistochemical staining and cytogenetic evaluation. 6 Molecular studies have demonstrated the presence of the chromosomal translocation t(X;18)(p11;q11) in over 90% of cases of SS. This anomaly leads to a hybrid product which involves gene SYT on 18p11 and one gene of SSX family on chromosome X, mostly SSX-1, less frequently SSX-2, and seldom SSX-4. 7 For differential diagnosis, metastatic sarcoma should be considered as well as sarcomatoid renal cell carcinoma and hemangiopericytoma which may have similar histological features. 8 We describe a case of primary SS of the kidney, and present a review of the relevant literature.
Case Report
A 33-year-old male presented with a history of persisting right flank pain which started a 2-month before and before that intermittedly occurred in the last year. A 12 cm mass is detected on ultrasonography in the middle of the kidney at corticomedullary junction. Clinically, xanthogranulomatous nephritis is suspected as the patient meanwhile represented with signs of inflammation like fever, pain and high laboratory levels of inflammatory parameters in serum. Low density mass is described in computed tomography (CT) images. CT-guided biopsy and frozen section were made, which revealed pyelonephritis with spindle cell tumor of uncertain malignancy. The tumor part was too little, to be histologically differentiated. Patient was scheduled for open exploration of the right kidney (Figure 1A). Pre- and post-operative serum creatine was normal (84 umol/L). Intraoperatively, the kidney demonstrated with macroscopic necrosis that shined through the renal capsule. At two locations frozen section were taken, demonstrating inflammation and necrotic tissue. The necrosis was evacuated in order to drain the kidney, but finally and corresponding to the abdominal CT scan, the central part of the kidney was necrotic and renal pelvis was open, the preoperatively implanted DJ-ureteral catheter laid in the necrotic central part of the kidney. Consecutively, right nephrectomy was applied, which showed a 12×7×5 cm tumor (Figure 1B) with vessel invasion. The tumor showed grayish white tissue with multiple blood spaces, necrosis and cysts. Pathologic confirmation was performed by immunohisto-chemical methods and in-situ-hybridization (FISH). Histologic examination of tumoral tissue, stained with hematoxylin and eosin, revealed solid cellular nests of monomorphic spindle cells with non-uniformly bounded cytoplasm in large areas and fascicles with cystic structures settled among them (Figure 1B-D). For the immunohistochemistry deparaffinized and rehydrated FFPE tissue sections (1-2
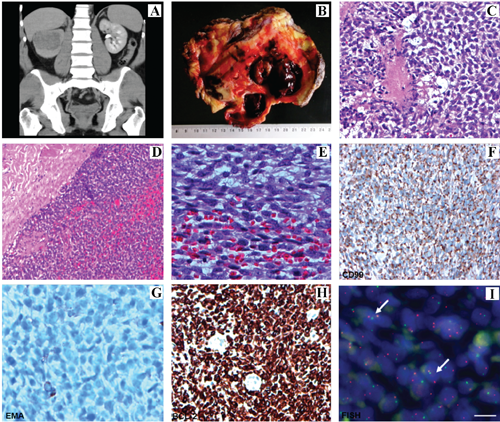
A) Open lumbal nephrectomy; B) nephrectomy showing a 12×7×5 cm tumor; C-E) histologic examination of tumoral tissue (hematoxylin and eosin); F-I) immunohistochemical examination of the tumor (CD99; EMA; BCL-2; FISH respectively).
Immunohistochemistry was performed with an aim to detect the nature of the tumor cells and to differentiate the tumor and excluding the differential diagnosis which in these cases include sarcomatous renal cell carcinoma, nephroblastoma of the adult type, spindle /mesenchymal tumor of the kidney, dedifferentiated liposarcoma, rhabdomyosarcoma, leiomyosarcoma and neuroendocrine tumors. The tumor cells were positive for vimentin, CD99, BCL-2 and focally positive for AE1/AE3 and EMA. There was no reaction to CEA, desmin, chromogranin, synaptophysin, enolase, S-100, CD-10, CD-117, Caldesmon, bcl-2, β-catenin, PAX2, and WT-1. The cysts were lined by epithelium, which was focally positive for AE1/AE3 and for PAX2. In the molecular study of the study, there is sure rearrangement in the SYT-SSX gene (Figure 1E-I). According to these findings a diagnosis of monophasic spindle cell synovial sarcoma was done. CT imaging in April 2014 demonstrated no signs of metastatic disease. Patient is scheduled for adjuvant chemotherapy with 4 cycles of adriamycin and ifosfamid.
Discussion
Primary renal synovial sarcoma (RSS) is rare, constitutes a subtype of cases identified as embryonal sarcoma of the kidney. There is currently no standard therapy because of the limited number of reported cases. Among sarcomas of the kidney, leiomyosarcoma is the most frequent type compromising 40-60%, followed by rhabdomyosarcoma, chondrosarcoma, liposarcoma, angiosarcoma, hemangiopericytoma, and osteosarcoma. 9
It affects both genders and young individuals with a range of 20-50 years of age. 10 The limited number of cases reported has shown a gender ratio close to one, a mean age at diagnosis of 37 years (ranging between 13 and 67), and mean tumor diameter of 11 cm, ranging from 3 to 21 cm. The rate of metastasis on admission seems to be low. 10
Making an accurate diagnosis is always a problem, since renal synovial sarcoma is usually confused with other soft-tissue sarcomas under the microscope, due to its rarity and similar presentation compared to other renal tumors, the diagnosis with the findings of clinical presentation and imaging modalities is difficult. The macroscopic presentation with solid and cystic areas are the same as sarcomatours renal cell carcinoma, others sarcomas and Wilm's Tumor. 11
Poorly-differentiated SS is composed of sheets of undifferentiated round cells with hyperchromatic nuclei and frequent mitoses, and shows the poorest outcome. 12 Biphasic SS can be diagnosed with the presence of both epithelial and spindle cell components. On the other hand, the monophasic subtype comprises only an epithelial or spindle cell component. However, monophasic SS may be difficult to differentiate from other spindle cell sarcomas. 10 The intratumoral cystic areas were lined by epithelium which was focally positive for pancytokeratin (AE1/AE3) and for PAX-2 which may represent only entrapped tubuli, as early reported by Karaffin et al. 13
Through reviewing the literature, we found multiple immunohistochemical markers that have been investigated in cases of RSS. Markers such as Bcl-2, vimentin, CD99, EMA, CD56, S100, desmin, SMA, CD34, AE1/AE3, and WT-1 have all been tested for the use in diagnosis of the cancer from paraffin-embedded sections.
WT-1 expression is always found in cases of adult Wilms’ tumor but not in primary RSS. Furthermore, malignant peripheral neuroectodermal tumor (MPNET) is typically positive for NSE and approximately 50% to 70% of tumors express S100, while primary renal synovial sarcomas are negative for both. 14
The current diagnostic gold standard for synovial sarcoma is to demonstrate the fusion of the SYT (Synonyms: SS18-synovial sarcoma translocation, chromosome 18) gene on chromosome 18 to either SSX1 (synovial sarcoma, X breakpoint 1) or SSX2 (synovial sarcoma, X breakpoint 2) gene on chromosome Xp11.7,8 Previous work from other colleagues such as Lopes et al. 15 and others could not detect the gene rearrangement.
SS is considered to be an aggressive form of STS, with a high probability of systemic spread, and it is considered to be more sensitive to chemotherapy than other soft tissue sarcomas (up to 53% of response rate). 16 Based on this observation, SS are often treated aggressively with multimodal therapies.
Conclusions
In conclusion, primary renal SS is an extremely rare neoplasm. Histomorphological and immunohistochemical features may be related to other renal spindle cell cancers and differential diagnosis is difficult. FISH-analysis is the most rapid way with reasonable price and the most accurate method for its diagnosis. In comparison with other authors who reported this rare tumor, we can detect the rearrangement in the SYT-SSX-Gene which supports the diagnosis. Although the surgical resection and ifosfamide-based chemotherapy regimen are accepted treatment protocols for the management of renal SS, the prognosis is poor, but may be the monophasic subtype has better prognosis which highlights the importance of the correct, early diagnosis and good differentiation between sarcomatous renal cell carcinoma, other spindle/round cell tumors of the kidney and primary synovial sarcoma of the kidney with special highlighting of the importance to differentiate morphologically and with additive molecular methods between monophasic and biphasic synovial sarcoma. Adjuvant chemotherapy in this setting delivers a 8-10% overall survival benefit.