
Abstract
BACKGROUND:
Hereditary renal cell carcinoma (RCC) is a complex and rapidly evolving topic as there is a growing body of literature regarding inherited syndromes and mutations associated with an increased risk of RCC.
OBJECTIVES:
We sought to systematically review 13 hereditary syndromes associated with RCC; von Hippel-Lindau Disease associated RCC (VHLRCC), BAP-1 associated clear cell RCC (BAPccRCC), Familial non-von Hippel Lindau clear cell RCC (FccRCC), Tuberous Sclerosis Complex associated RCC (TSCRCC), Birt-Hogg-Dub
RESULTS:
Hereditary RCC is generally associated with an early age of onset, multifocal and/or bilateral lesions, and aggressive disease course. VHLRCC, BAPccRCC, FccRCC, and certain mutations resulting in SDHRCC are associated with clear cell RCC (ccRCC). HPRCC is associated with Type 1 papillary RCC. HLRCC is associated with type 2 papillary RCC. BHDRCC is associated with Chromophobe RCC. TSCRCC, PHTSRCC, MiTFtRCC, TFEBRCC, ADPKDRCC, certain SDHRCC and ALKRCC have variable histology.
CONCLUSIONS:
There has been tremendous advancement in our understanding of the pathophysiology of hereditary RCC. Ongoing research will refine our understanding of hereditary RCC and its therapeutic targets.
Keywords
INTRODUCTION
Hereditary forms of renal cell carcinoma (RCC) comprise up to 5% of RCC cases. In patients presenting with metastatic RCC (mRCC) and uncommon RCC variants, the prevalence of a germline mutation was estimated at 16% and 20%, respectively [1]. General features common to most hereditary forms of RCC are early age of onset, multifocal and/or bilateral lesions, and aggressive disease course. There are a growing number of cancer syndromes and predisposing mutations without identified syndromes that have been connected to RCC. Mammalian target of rapamycin (mTOR) and hypoxia-inducible factor (HIF)-2 alpha (HIF-2α) are an established link in the phosphoinositide 3-kinase (PI3K)-protein kinase B (AKT)-mTOR-HIF-erythropoietin (EPO) axis that plays a vital role in hereditary RCC (Fig. 1) [2–4]. The treatment landscape for clear cell RCC (ccRCC) has evolved rapidly over the last few years. In August, 2021, the Food and Drug Association (FDA) approved the HIF-2α inhibitor belzutifan for cancers associated with von Hippel Lindau Disease (VHL), including VHLRCC [5]. Table 1 outlines the key characteristics of heritable RCC and potential therapeutic targets and clinical trials.
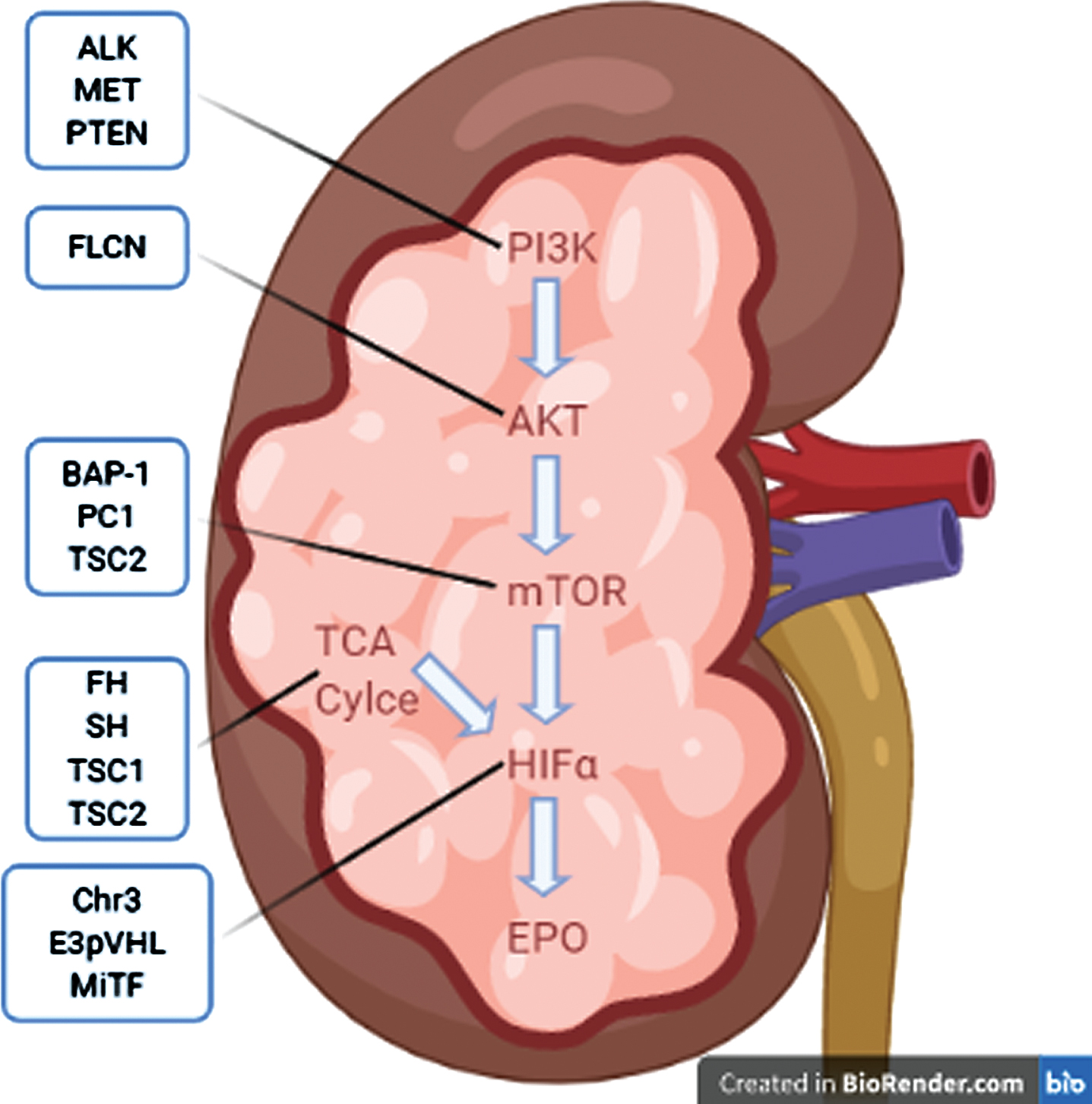
Summary hereditary RCCs via common pathway of PI3K-AKT-mTOR-EPO. Abbreviations: Phosphoinositide 3-Kinase (PI3K), Protein Kinase B (AKT), Mammalian Target of Rapamycin (mTOR), Hypoxia-Inducible Factor Alpha (HIFα), Tricarboxylic Acid (TCA), Erythropoetin (EPO), Tyrosine Kinase Met/Hepatocyte Growth Factor Receptor (MET), Phosphatase and Tensin Homolog (PTEN), Anaplastic Lymphoma Kinase (ALK), Folliculin (FLCN), Ubiquitin Carboxyl-terminal Hydrolase BAP1 (BAP1), Polycystin 1 (PC1), Fumarate Hydratase (FH), Succinate Dehydrogenase (SDH), Tuberous Sclerosis Complex-1 and 2 (TSC1 and TSC2), other Chromosome 3 alterations (Chr3), E3- Von Hipple Lindau protein (E3pVHL), Melanocyte Inducing Transcription Factor (MiTF). Image created using BioRender.com.
Summary of hereditary RCC with gene, loci based on OMIM database, typical histology, therapeutic targets, and ongoing clincial trials
Herein, we review hereditary RCC; von Hippel-Lindau Disease associated RCC (VHLRCC), BAP-1 associated clear cell RCC (BAPccRCC), Familial non-von Hippel-Lindau clear cell RCC (FccRCC), Tuberous Sclerosis Complex associated RCC (TSCRCC), Birt-Hogg-Dub
CLEAR CELL PREDOMINANT HERITABLE RCCs
von Hippel-Lindau disease associated RCC (VHLRCC)
VHL is AD with an incidence of approximately 1 in 30,000. VHL is characterized by mutations of VHL, found at loci 3p25, and a predisposition to various tumors including hemangioblastoma of the central nervous system and retina, pheochromocytoma, and RCC [6]. The VHL gene expresses the protein VHL (pVHL), which forms a complex with elongin B, elongin C, Rbx1 and cullin 2, thus forming the pVHL-E3 ubiquitin ligase complex (pVHLE3) [7–10]. This complex then ubiquitinates HIF-α, leading to its degradation [8, 11–15].
RCC develops in about 25–45% of patients with VHL, with age of onset during 2nd to 4th decade of life and by age 60, penetrance is ∼70% [16]. VHLRCC is typically ccRCC, multifocal and bilateral [17].
Understanding of the pVHLE3-HIFα-EPO pathway ushered in the use of multi-kinase targeted therapies which have transformed the therapeutic landscape in ccRCC [18]. Multi-kinase inhibitors alone and in combination with checkpoint inhibitors form the backbone of ccRCC treatment.
HIF-2α is a critical therapeutic target in ccRCC, and the advent of HIF-2α inhibitors has further revolutionized the treatment for RCC. Belzutifan (MK-6482), a HIF-2α inhibitor, showed activity in a phase II trial in patients with localized VHLRCC in which 53 out of 61 patients had response [5]. In another phase I/II trial in advanced RCC with 1 prior line of treatment, belzutifan resulted in tumor response in 35 of 52 patients and was recently granted FDA approval [19].
In VHLRCC patients, sunitinib did not show efficacy for hemangioblastomas, but pazopanib demonstrated response in VHL lesions other than VHLRCC [20, 21].
BAP-1 associated clear cell RCC (BAPccRCC)
BAPccRCC is an inheritable form of ccRCC associated with BAP-1 mutation; loss of BAP-1 has been demonstrated to indirectly upregulate mTOR [22]. In sporadic ccRCC, a common pathway in tumorigenesis involves an initial mutation to VHL followed by a mutation to BAP-1 or PBRM1 followed by a third mutation of the remaining wildtype BAP-1 or PBRM1, a gene that encodes SWI/SNF chromatin remodeling complex found in 41% of ccRCC [23, 24]. Inherited BAPccRCC was found in 83 unrelated patients with unexplained familial RCC [22]. Tumors with BAP-1 and PBRMI loss were associated with rhabdoid features [25]. BAP-1 and PBRM1 mutations may define subtypes of ccRCC with poorer prognosis for BAP-1 subgroup [23]. BAP-1 loss in in vitro models sensitized cells to ionizing radiation and poly adenosine diphosphate-ribose polymerase (PARP) inhibitors are being studied in ongoing trials (NCT03786796, NCT03207347) [25].
Familial non-von Hippel-Lindau clear cell RCC (FccRCC)
FccRCC is AD and characterized by inherited ccRCC without evidence of VHL [26, 27]. Mutations to various genes via chromosome 3 translocations, excluding those to VHL, MET or CUL2, can account for these cases [28]. In a review of 60 kindreds with at least 2 patients with ccRCC, there were 145 affected patients with familial RCC without another syndrome or mutation [28]. The mean age of onset was 53 years, which is younger than sporadic cases but older than VHLRCC; 14/145 individuals had multiple tumors at diagnosis or developed a second primary and 17/145 patients developed an additional benign or malignant neoplasm, but there was no evidence to suggest inherited susceptibility.
NON-CLEAR CELL PREDOMINANT HERITABLE RCCs
Tuberous sclerosis complex (TSC) associated RCC (TSCRCC)
TSCRCC is an AD disease characterized by seizures, cognitive impairment, hamartomas in various organs and RCC [29]. Mutations of TSC1 and TSC2, loci 9q34 and 16p13 respectively, result in this syndrome [30, 31]. TSC1 encodes Hamartin and TSC2 encodes Tuberin, which heterodimerize to produce a complex that ultimately inhibits the mTOR pathway [32, 33]. The incidence of TSCRCC is similar to the incidence of RCC in the general population, except with earlier age of onset and pathological heterogeneity [34, 35]. Renal Angiomyolipoma (RAML) and atypical RAML are also associated with TSC [36, 37].
Due to signaling through mTOR and HIF, mTOR and HIF inhibitors are potential therapies. Everolimus resulted in durable response in TSC2 mutant RCC [38]. In the EXIST-2 trial, a phase 3 trial of 118 patients with at least one 3 cm or larger angiomyolipoma and a diagnosis of tuberous sclerosis or sporadic lymphangioleiomyomatosis treated with everolimus or placebo, everolimus demonstrated 42% response rate in angiomyolipomas [39]. Four year follow up of EXIST-2 with median duration of everolimus exposure of 46.9 months demonstrated a renal lesion reduction in 97% of patients [40]. There is an ongoing phase II clinical trial in solid tumor patients with mutant TSC1 or TSC2 using everolimus (NCT02201212).
Birt-Hogg-Dubé syndrome associated RCC (BHDRCC)
BHD is as an AD syndrome characterized by fibrofolliculomas of the skin, spontaneous pneumothorax, and increased risk of RCC [41–44]. The FLCN gene, loci 17p11.2 [45], encodes for the protein FLCN that functions to regulate cell metabolism in conjunction with Folliculin-Interacting Protein (FNIP) 1 and 2. This regulation occurs through the AMPK energy sensing, mTOR proliferation cascade, and Ppargc1a mitochondrial oxidation [46–52]. In a review of 89 individuals from 51 families with BHD mutations, 34% of individuals had RCC, with median age of onset around 50 years [53]. The predominant histologies are Hybrid oncocytic/chromophobe tumors (HOCT) and chromophobe RCC [41, 54]. These tumors are generally slow growing and can be managed surgically [55]. Everolimus showed response as 6th line therapy in a patient with BHD-RCC [56].
PTEN hamartoma tumor syndrome associated RCC (PHTSRCC)
PHTS encompasses AD conditions caused by germline mutation of PTEN, including Cowden Syndrome [57] and Bannayan-Riley-Ruvalcaba Syndrome [58]. PTEN, loci 10q23, encodes for the protein Phosphatase and tensin homolog [59, 60], which blocks PIP3 dependent PI3K activation of AKT [61–64]. In a series of 219 patients with a germ line PTEN mutation, 9 were found to have RCC, with 6 being papillary and 2 being chromophobe [65]. RECORD-3 was a phase IIb clinical trial comparing everolimus to sunitinib in patients with advanced RCC that did not meet its primary outcome [66]. However, subsequent analysis of based on PTEN expression found longer PFS (10.5 months vs 5.3 months, p < 0.001) in patients with PTEN loss vs not, respectively, for those treated with everolimus. There was no significant difference in PFS (10.3 months vs 10.9 months, p = 0.475) based on PTEN expression in those treated with sunitinib [67]. This evidence suggests that decreased PTEN expression, as would be seen in PHTS, would derive more benefit from everolimus compared to sunitinib.
Microphthalmia-associated transcription family (MiTF)translocation RCC (MiTFtRCC)
MiTFtRCC is caused by AD germline mutation of one of the MiTF transcription factors, which are: MITF, TFE3 loci Xp11.23, TFEB loci 6p21.1, and TFEC [68–72]. There are 2 further subgroups of MiTFtRCC defined by the presence of rearrangement of TFE3 via Xp11 and TFEB via t(6;11) [73]. TFE3 and TFEB function in the regulation of autophagy induction in response to DNA damage and starvation [74]. MITF mutation MiE318 has been linked to RCC and melanoma, with mutation carriers having a 5 times higher risk of melanoma, RCC or both [75]. Xp11 translocation RCC causes 20–40% of childhood RCC and 1–4% of adult RCC with age of onset around 50 years [76, 77]. The presence of ASPSCR1-TFE3 fusion has a poorer prognosis than other fusions [78]. These tumors have cells with clear to eosinophilic cytoplasm, often mixing features of clear cell and papillary RCC [79]. t(6;11) RCC is quite rare with an age of onset of 30 years and poor prognosis [79–81]. These RCCs usually have weak expression of epithelial markers on IHC, sometimes express melanoma markers, and may not respond well to VEGF targeted therapy, so a misclassification as clear cell may lead to unexpectedly poor outcomes [54]. There is an ongoing phase II trial to evaluate the efficacy of axitinib plus nivolumab in patients with TFE/translocation RCC (NCT03595124).
RCC with chromosome 6p (TFEB) amplification (TFEBRCC)
TFEBRCC is related to MiTFtRCC because it has an amplification or rearrangement of the TFEB gene other than t(6;11). The unique features to this category are age of onset about 65 years old, areas of high grade epithelioid cells with pseudo or true papillary histology, and more aggressive course [82–84]. These patients have a more aggressive course, including more likely to present with advanced disease and to metastasize [82]. The series by Gupta et al demonstrated 24 cases with associated amplification of VEGFA with 11 (46%) of patients having metastasis and death from RCC [84].
Autosomal dominant polycystic kidney disease (ADPKD) associated RCC (ADPKDRCC)
The association of ADPKD and RCC has been a controversial topic over the years. ADPKD is caused by mutations of PKD1 at loci 16p13.3, which encodes for the transmembrane protein polycystin-1, and PKD2 at loci 4q21.1, which encodes for the transmembrane cation channel polycystin-2, with polycystin-1 regulating the function of polycystin-2. Approximately 85% of ADPKD cases are due to mutant PKD1 and 15% due to mutant PKD2 [85]. ADPKD is characterized by cysts of the kidneys, liver, pancreas, seminal vesicles, and meninges and connective tissue disorders affecting heart and vasculature [85–87]. Mutations of PKD1 are associated with more renal cysts and onset of renal failure about 20 years earlier [88, 89].
The RCC found in patients with ADPKD has unique characteristics. In a case series of 25 cases of RCC in ADPKD (3 new cases from the Mayo Clinic and 22 cases from literature review), there was no increased risk of RCC in those with ADPKD compared to historical controls. However, the following characteristics were more common in ADPKD than the general population of RCC patients: presence of a triad of fever, flank pain, weight loss, concurrent bilateral or multifocal tumors, and sarcomatoid histology [90, 91]. A more contemporary retrospective review of patients with ADPKD included 240 pts with a median age of 54 years that underwent nephrectomy and 5% were incidentally found to have RCC with the majority being papillary histology [92]. A review of renal transplant patients in the US of 10,626 patients with PKD, including ADPKD, found an increased risk of RCC compared to the general population but did not find ADPKD to be a risk factor for developing RCC [93].
One potentially confounding factor in ADPKDRCC is hemodialysis and its impact on the incidence of RCC. It is known that the majority of patients requiring chronic hemodialysis, regardless of the etiology, eventually develop cystic kidney disease. The presence of this acquired cystic kidney disease increases their risk of developing RCC by 3–6 times over historical general United States population risk from the Mayo Clinic [94]. It has also been shown that depending on the types of renal injuries, patients may have a predilection to a specific type of RCC [95]. A retrospective analysis of patients with ADPKD who did not have end stage renal disease in Taiwan demonstrated a higher incidence of liver, colon and renal cancers in those with ADPKD as compared to age matched controls [96]. This supports the theory that ADPKD, not just associated hemodialysis, increases the risk of RCC. Although the mechanism is thought to be mediated via the mTOR pathway, clinical studies have not yet demonstrated efficacy of mTOR inhibitors for treatment. It is possible that promising preclinical dual mTOR/PI3K inhibitors will demonstrate efficacy in the treatment of ADPKD and/or ADPKDRCC[97].
Hereditary leiomyomatosis associated RCC (HLRCC)
HLRCC is an AD disease characterized by uterine and skin leiomyomas and RCC [98]. Mutations of Fumarate Hydratase (FH) gene, found at loci 1q42-q44, account for these cases. The enzyme fumarate hydratase catalyzes the conversion of fumarate to malate in the Krebs cycle [99]. In a study of 21 families with HLRCC, renal tumors occurred in 13/21 families and while various histologies were seen, tumors had amphophilic cytoplasm and large nuclei with large inclusion-like nucleoli, making them similar to type II papillary RCC [98].
The RCC cells of HLRCC depended on glucose for adenosine triphosphate generation via aerobic glycolysis, known as the Warburg effect, due to a defect in the Krebs cycle. Increased oxidative stress and/or increased fumarate lead to increased levels of HIF, which results in increased VEGF [100]. The AVATAR trial (NCT01130519) is a phase II study in HLRCC or sporadic papillary RCC using a combination of bevacizumab and erlotinib and demonstrated median PFS of 21.1 months in the HLRCC cohort and 8.7 months in the sporadic cohort [101]. Another clinical trial (NCT02495103) using vandetanib and metformin in HLRCC or SDH associated RCC or sporadic papillary RCC has completed and results are pending. Vandetanib, an inhibitor of vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and rearranged during transfection (RET), is used to treat medullary thyroid cancer [102]. A retrospective review of 10 HLRCC patients in Korea showed clinical benefit with bevacizumab plus erlotinib in 9 patients with PFS 13.3 months and overall survival of 14.1 months [103].
Succinate dehydrogenase RCC (SDHRCC)
SDHRCC is an AD disease characterized by RCC with or without pheochromocytoma and paraganglioma [104, 105]. The 4 genes that encode for mitochondrial SDH are SDHA at loci 5p15 [106], SDHB at loci 1p36 [107], SDHC at loci 1q21 [108], SDHD at 11q23 [104] and this syndrome is known to occur in all except SDHA. When SDH is dysfunctional, succinate accumulates in the mitochondria and is transported into the cytosol where it inhibits HIFα prolyl hydroxylases which leads to stabilization and activation of HIFα [109]. SDHRCC has been described in patients with mutation of SDHB, SDHC, and SDHD. SHDBRCC was first described in 2004 in a kindred study of 2 families in which there was RCC diagnosed with age of onset < 30 years old and variable histology with solid, clear cell, and cells with granular eosinophilic cytoplasm [110]. In this study, the prevalence of RCC in SDHB mutation carriers was 5–10%. The unique histology of SDHBRCC includes nests of cuboidal tumor cells with granular or bubbly cytoplasm and prominent cytoplasmic inclusions that correspond to giant mitochondria [111, 112]. SDHCRCC and SDHDRCC typically have a later age of onset, in the 40’s, and clear cell histology as compared to SDHCRCC [113].
In SDHRCC, as in HLRCC, treatments that interact with cellular metabolism may be of value. There is on ongoing trial of pan VEFG-inhibitor, vandetanib, plus metformin in patients with HLRCC or sporadic papillary carcinoma (NCT02495103). There is also a phase II trial of guadecitabine, a DNA methyl transferase inhibitor in patients with SDHRCC (NCT03165721). There is a trial of talazoparib, a PARP inhibitor [114] and avelumab, an anti PD-L1 antibody [115], in patients with VHL, FH or SDH deficiency (NCT04068831). The compound CB-839, a glutaminase inhibitor is also under investigation in a trial including SDHRCC patients (NCT02071862). A phase II trial is underway to evaluate cabozantinib, a VEGF-inhibitor, and nivolumab, a PD-1 inhibitor, in patients with non-clear cell RCC including SHDRCC and HLRCC (NCT03635892).
Hereditary papillary RCC (HPRCC)
HPRCC is an ADRCC disease characterized by familial papillary RCC. Mutations in the MET gene, found at loci 7q31-34 [116], account for this disease. HPRCC tumors demonstrate type I papillary architecture, first described in a kindred study of 3 families with hereditary papillary RCC [117]. Age of onset is variable. In a study of 129 sporadic papillary tumors, MET mutations were found in only 17 cases, demonstrating that non hereditary papillary RCC is likely driven by other genetic alterations [118].
Due to the role of MET in HPRCC and chromosome 7 trisomy in sporadic papillary RCC, MET inhibitors have the potential to make a significant advancement in treatment. There are 2 clinical trials with MET inhibitors in papillary RCC (NCT00726323, NCT02019693). A phase II study of foretinib, inhibitor of MET, VEGF, RON, AXL, and Tie-2, in patients with papillary RCC included 10 patients with germline MET mutation and found that response rates were higher in patients with MET mutations compared to those without [19].
ALK-rearrangement RCC (ALKRCC)
ALKRCC is characterized by various Anaplastic Lymphoma Kinase (ALK) fusion proteins, including VCL-ALK in patients with sickle cell trait [119, 120], TPM3-ALK [121], STRN-ALK [122]. ALK, loci 2p23.2, is a receptor tyrosine kinase of the insulin superfamily [123, 124]. With various fusion partners, ALK has been reported in many malignancies and is a relatively new entity in RCC. These cases have varied histopathological features and comprises < 1% of RCC [125]. Due to the presence of ALK fusion, this group of RCC may be responsive to ALK inhibitors. CREATE is an ongoing phase II basket study with crizotinib in solid tumors with activating alterations to ALK or MET (NCT01524926). Another phase II basket trial is testing entrectinib in solid tumors with NTRK1/2/3, ROS1, or ALK gene fusion that is ongoing as well (NCT02568267).
CONCLUSION
The identification of cases of RCC that shared unusual characteristics such as early age of onset, other syndrome features, atypical histologies, and focal versus multifocal presentation led to identification of genes with oncogenic potential. This has revolutionized our understanding of RCC and more patients with inherited susceptibility to RCC are being identified as our knowledge expands. The next step is connecting the mutant genes to their aberrant gene products and related pathways. This will provide understanding that can not only be applied to RCC but also across scientific specialties to further our understanding of the various pathways. The final step is to discover targeted therapeutics tailored to the specific aberrant pathway(s) in each patient’s cancer. Ongoing clinical trials with known targeted therapeutics along with ongoing drug discovery are critical to our continued advancement and goal of personalized medicine in future.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgements.
FUNDING
The authors report no funding
AUTHOR CONTRIBUTIONS
Conceptualization, S.J.D. and S.G; writing—original draft preparation, S.J.D.; writing—review and editing, S.J.D. and S.G.; visualization, S.J.D.; supervision, S.G.; project administration, S.G. All authors have read and agreed to the published version of the manuscript.
CONFLICT OF INTEREST
Scott J. Dawsey and Shilpa Gupta have no conflict of interest to report.