
Abstract
Birt-Hogg-Dubé syndrome, an extremely rare genetic disorder, is characterized by the development of fibrofolliculomas, lung cysts and subsequent recurrent pneumothorax, and kidney neoplasia. This report highlights the case of a 56-year-old female with a history of right vestibular schwannoma status post stereotactic radiotherapy and vulva bartholin’s gland carcinoma who was initially evaluated by primary care for a 6-month history of intermittent, red, raised, widespread rash accompanied by fever, chills, and body aches. A punch biopsy of the rash was performed, which was notable for an urticarial tissue reaction with focal changes of leukocytoclasia and negative direct immunofluorescence. Laboratory tests, which included an autoimmune genetic and periodic fever panel, were unremarkable. Whole genome sequencing returned positive for a pathogenic variant in folliculin gene, consistent with a diagnosis of Birt-Hogg-Dubé syndrome.
Introduction
Birt-Hogg-Dubé syndrome (BHDS) is a rare autosomal dominant disorder that presents typically with dermatosis, commonly fibrofolliculomas, and predisposes affected patients to an increased risk of developing pulmonary cysts, spontaneous pneumothorax, and renal cancer. 1 The genetic basis for BHDS involves a germline insertion/deletion and nonsense mutation in the FLCN gene, and there is currently no definitive estimate of the prevalence of the disease.1,2
Skin lesions are typically the initial symptoms, usually appearing in the third and fourth decade of life. 2 However, fibrofolliculomas can also be present in the older age group. Disease presentation and severity can vary significantly, and this further contributes to the diagnostic challenge BHDS poses in the clinical setting.
Studies have confirmed that spontaneous pneumothoraces and renal tumors are the two major extracutaneous manifestations of BHDS. 2 The diagnosis of BHDS should prompt screening for pulmonary cysts and renal neoplasm at the very least. Management is largely conservative with the aim of addressing organ-specific symptoms.
Case presentation
We present the case of a 57-year-old female with a past medical history of osteoarthritis, right vestibular schwannoma status post stereotactic radiotherapy, vulva Bartholin’s gland carcinoma, and hypothyroidism who initially presented to primary care with a complaint of “hive-like” skin rash in the lower abdominal, pubic, and medial thigh areas that waxed and waned for about 6 months (Figure 1). Associated symptoms include fever, myalgia, and fatigue. The rash does not itch, and she has no known allergies. Symptoms typically occur every 1–3 weeks and resolve after 2–4 days. However, prior to presentation, frequency of symptoms has increased to every 4 days. Myalgia heralds the onset of skin rash and is followed by fever that could be as high as 102 F. She denies any recent exposure to allergens nor changes to skin care and detergent products. This was diagnosed as Urticaria, and the patient was sent home on a 5-day course of Steroid. Three weeks later, the patient returned to the primary care clinic with worsening symptoms and spreading of the rash to face and scalp. At this visit, a diagnosis of Chronic Papular Urticaria was made, and she was placed on Cimetidine and topical triamcinolone. Her symptoms became more severe in frequency and distribution 2 weeks later. Laboratory evaluation was only significant for elevated CRP—11.8 mg/L (0.0–8.0 mg/L).

Diffuse red raised papule with urticarial-like morphology on lower abdomen/pubic (left) and right lateral thigh region (right).
A dermatology referral was made, and two series of skin biopsy were performed. The first biopsy showed urticarial tissue reaction with focal leukocytoclastic changes and negative direct immunofluorescence (Figure 2). Repeat punch biopsy a week later showed nonspecific findings of perivascular lymphocytic infiltrates with a few scattered eosinophils, mast cells, and neutrophils. Direct immunofluorescence did not reveal evidence for a specific dermatosis. Serological tests including Complement, dsDNA Antibodies, Antibody to Extractable Nuclear Antigen, ANA, Creatine Kinase, ESR, and Anti-CCP were normal. CBC, CMP, and LFT were unremarkable. CRP was elevated at 50.4 mg/L. Urine studies were also within normal limit. Rheumatology was involved, and on review, Chronic urticaria index and anti-TPO antibodies were evaluated. Both tests returned normal. Autoinflammatory Primary Immunodeficiency Gene Panel (CARD14, IL10RA, IL10RB, IL1RN, IL36RN, ISG15, LPIN2, MEFV, MVK, NLRP12, NLRP3 (CIAS1), NOD2 (CARD15), PLCG2, PSMB8, PSTPIP1 (CD2BP1), RBCK1 (HOIL1), SH3BP2, and TNFRSF1A) and Serum protein electrophoresis also returned negative. Pelvic ultrasound showed no evidence of endometrial, myometrial, or adnexal solid or cystic mass. At this point, referral was made to clinical genomics for full genetic work-up. The patient was interested in finding answers for her symptoms, and given personal history of cancer, whole genome sequencing (WGS) was considered the most appropriate option. WGS revealed the presence of heterozygous mutation in the FLCN gene with pathogenic variant c.779+1G>T reported to cause autosomal dominant BHDS.

Histology of right lower abdomen skin punch biopsy showing predominantly perivascular lymphocytic and eosinophilic inflammation with focal perivascular and interstitial neutrophils on background of ectatic small vessels, and mild acanthosis with focal vacuolar interface degeneration more consistent with an urticarial reaction.
Imaging of Chest and abdomen were conducted. CT chest showed small lung cysts, calcified granulomas and noncalcified pulmonary micronodules (Figure 3). Abdominal MRI showed a 14-mm lesion in the left kidney, which demonstrated mild enhancement, likely representing a hemorrhagic or proteinaceous cyst. There were several additional renal and hepatic cysts with no concerning features (Figure 4). She required no immediate intervention at time of diagnosis and continued to follow-up with pulmonology, urology, and dermatology on a regular schedule. She was advice to minimize activities that can precipitate a spontaneous pneumothorax such as scuba diving, high altitudes and flying. Genetic counseling and testing were offered to family members.
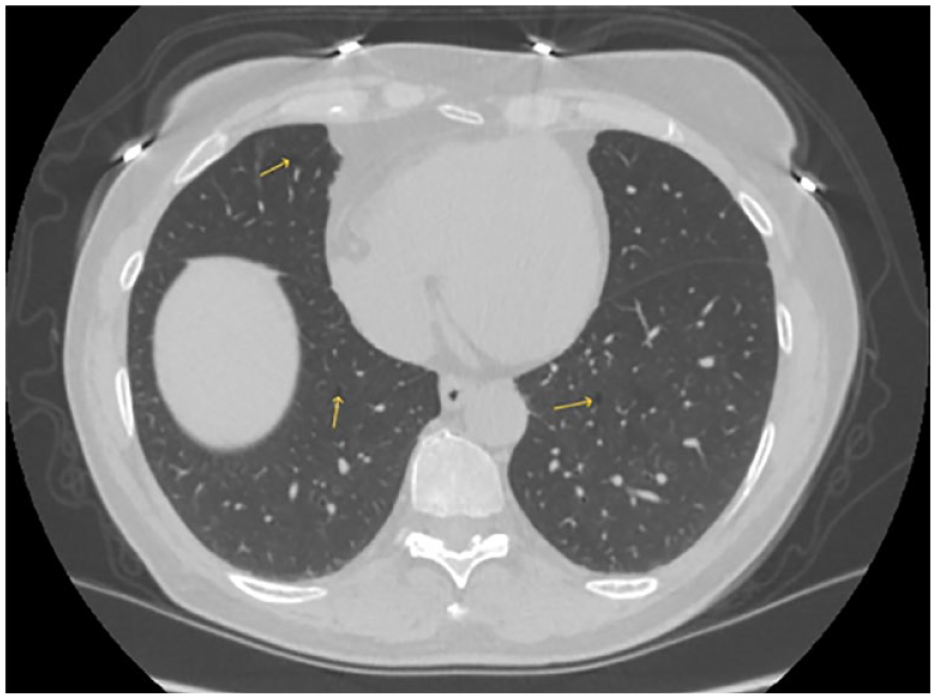
Small areas of cystic lesions scattered across both lungs (yellow arrow).
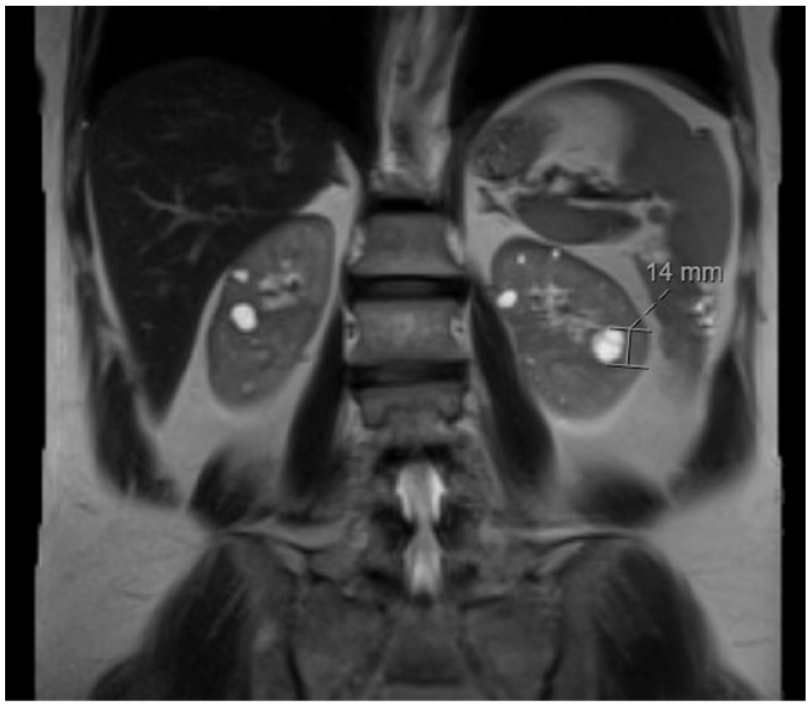
Mildly enhanced left kidney lesion with multiple bilateral renal cysts and some hepatic cysts.
Discussion
The earliest probable account of BHDS was reported in 1925 by Burnier and Rejsek in a 56-year-old female with skin-colored papules in head and neck region. Since then, multiple similar cases have been reported. However, familial pattern of the disease and extracutaneous manifestations was first reported by Hornstein and Knickerberg in 1975, hence the former name Hornstein-Knickerberg syndrome. 1 Drs. Birt, Hogg, and Dubé in 1977, while investigating a case of thyroid cancer with associated skin lesion like those described by Burnier and Rejsek, discovered the triad of fibrofolliculomas, trichodiscoma, and acrochordons as classic cutaneous manifestation of what is now known as BHDS. 1 Of the three, acrochordons are not diagnostic as they are prevalent in the general population and trichodiscomas are now considered remnant of scarred fibrofolliculomas.2,3
BHDS is known to be caused by germline mutation in the folliculin (FLCN) tumor suppressor gene on Chromosome 17p11.2. FLCN is expressed in major body tissues and interacts with folliculin interacting proteins 1 and 2 (FNIP1 and FNIP2) and produces a downstream modulating effect of the AKT-TOR signaling pathway via negative regulation of mammalian target of rapamycin.4–6 This mechanism is believed to be responsible for multisystemic manifestation and increased risk of malignancy associated with BHDS. 7 While over 150 mutations in FLCN gene have been identified in cases of spontaneous pneumothorax associated with BHDS, there is still no clear relationship between phenotypic manifestation in BHDS and specific variants of FLCN mutations. However, there has been observed increase in the number of lung cysts and consequently, recurrent spontaneous pneumothorax, in patients with Exons 9 and 12 mutations, while a deletion in polycytosine tract of exon 11 on the FLCN gene might be associated with lesser risk of renal cancer compared to other variants.8–10
Fibrofolliculomas are the most common cutaneous manifestation reported in up to 85% of BHDS cases. These are benign skin lesions presenting as small multiple flesh-colored, waxy yellowish papules typically on the upper face and neck region and manifesting between age 20 and 40 years old. It is important to note that skin findings might be absent in individuals less than 20 years of age.1,11 However, with increasing age, skin lesions are likely to involve other body parts including the trunk. Typical histopathologic finding in fibrofolliculoma is presence of circumscribed fibrosis with elongated fingerlike extension. 11 Perifollicular fibromas, angiofibroma and trichodiscomas have also been reported in a minority of BHDS cases. There have been case reports linking BHDS to melanoma, at least yearly skin exams are warranted with high attention to melanotic changes. During our literature review, no previous documentation of chronic neutrophilic urticaria as a dermatologic manifestation of BHDS was found.
Primary extracutaneous involvement in BHDS involves the development of pulmonary cystic disease. About 70%–80% of patients with BHDS go on to develop pulmonary cystic disease, which poses many challenges. Pulmonary cystic disease has not been linked to malignancy, but there is great morbidity in this condition as it puts patients at elevated risk for pneumothorax. Literature reviews show that recurrent spontaneous pneumothorax can often be a presenting symptom for the diagnosis of BHDS. 12 Patients with BHDS diagnosis should undergo CT chest and pulmonology evaluation on diagnosis. Patients should be advised to limit activities that put them at considerable risk for spontaneous pneumothorax, although there is a continuous debate on the need to restrict air travel in patients with multiple pulmonary cysts. 13 Renal manifestations of BHDS can also include cystic disease. However, the largest link with BHDS is renal neoplasm. Genetic analysis reveals that an upwards of 40% of patients with BHDS can develop renal neoplasms. 14 Renal cell carcinoma (RCC) is the most associated renal malignancy. Patients who have familial RCC should also be evaluated for BHDS as a potential genetic source. 15 As there is a great increase in the risk of RCC, urology follow-up is advised with yearly RCC screenings to allow for early detection.
The current guideline published by the European Birt-Hogg-Dubé consortium proposed major and minor criteria for the diagnosing BHDS. The major criteria include histologically confirmed fibrofolliculomas/trichodiscomas or Identification of FLCN pathogenic variant. Minor criteria are multiple lung cysts, renal cancer of mixed chromophobe and oncocytic patterns before age 50, and first-degree relative with BHDS. Patients with one major or two minor criteria meet the diagnostic threshold for BHDS.2,11,16 Negative FLCN gene testing does not exclude the diagnosis as up to 40% of patients with negative testing still meet diagnostic criteria. As presented in this case, our patient’s cutaneous manifestation of chronic neutrophilic vasculitis is not common for BHDS. Therefore, the first diagnostic clue of BHDS, which is often fibrofolliculomas, was absent. This made the diagnostic journey challenging, thus involving multispecialty input. Eventually, our patients met the major criterion of FLCN pathogenic variant and further investigation elicited minor criterion of multiple lung cyst on imaging.
The management of BHDS is targeted toward specific-organ system presentations. The skin lesions in BHDS are understood to be influenced by the downstream modulating effect of the AKT-TOR signaling pathway. As topical exosomes applications have been observed to improve skin quality, it is believed that the introduction of exosomes into BHDS skin lesions will disrupt the pathogenic signaling pathway and lead to improved collagen synthesis, thus alleviating some of the skin lesions. 17 There are also surveillance measures provisionally recommended for early detection of sequela of the disease process, especially renal cell cancer. Table 1 provides a comprehensive summary of these measures for patients with BHDS.
Management and surveillance of common organ-specific manifestation of patients with BHD syndrome.

BHDS has an autosomal dominance pattern of inheritance. Therefore, genetic testing is advised for offspring as the probability of inheriting the pathogenic variant is 50%. 11
Conclusion
While fibrofolliculomas are generally the most common dermatologic manifestation in BHDS, clinicians should be aware of other skin presentation such as urticarial vasculitis. The pattern of symptom manifestation may vary between patients. More so, extensive family history of neoplasm may also be absent in some cases. Due to the variability in phenotypic manifestation of the FLCN mutation variant, management and surveillance measures of patients diagnosed with BHDS need be individualized. There also needs to be ongoing discussion on refining diagnostic criteria for BHDS as new information about symptom presentation continues to be generated.
Footnotes
Acknowledgements
None.
Authors’ contributions
O.B. drafted the case presentation, introduction, and part of the discussion. He was also responsible for gathering images and figures used in the manuscript. D.S. drafted the abstract and some parts of the discussion. V.K. supervised the content and proofread manuscript for accuracy and completeness.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.