
Abstract
Oligomerization and aggregation of misfolded forms of α-synuclein are believed to be key molecular mechanisms in Parkinson’s disease (PD) and other synucleinopathies, so extensive research has attempted to understand these processes. Among diverse post-translational modifications that impact α-synuclein aggregation, glycation may take place at several lysine sites and modify α-synuclein oligomerization, toxicity, and clearance. The receptor for advanced glycation end products (RAGE) is considered a key regulator of chronic neuroinflammation through microglial activation in response to advanced glycation end products, such as carboxy-ethyl-lysine, or carboxy-methyl-lysine. The presence of RAGE in the midbrain of PD patients has been reported in the last decades and this receptor was proposed to have a role in sustaining PD neuroinflammation. However, different PD animal models demonstrated that RAGE is preferentially expressed in neurons and astrocytes, while recent evidence demonstrated that fibrillar, non-glycated α-synuclein binds to RAGE. Here, we summarize the available data on α-synuclein glycation and RAGE in the context of PD, and discuss about the questions yet to be answered that may increase our understanding of the molecular bases of PD and synucleinopathies.
INTRODUCTION
A common molecular denominator among synucleinopathies is the accumulation of aggregated forms of alpha-synuclein (α-synuclein) in the brain, which can be detected upon post-mortem examination. α-synuclein is abundantly expressed in the brain, but also in other tissues, where its role is less understood. However, the precise causative factors triggering the aggregation cascade that leads to oligomerization and fibrillization are not fully understood. It is believed that hydrophobic interactions involving membrane lipids may play a role in both oligomerization and aggregation [1]. Normally, neuronal α-synuclein is thought to interchange between monomeric and physiological oligomeric forms, with helical tetramers being considered a marker of possible “normal” oligomerization activity [2, 3]. Although still controversial, the destabilization of these tetramers is considered an initial step in aggregation of α-synuclein.
Post-translational modifications (PTMs) such as phosphorylation, acetylation, ubiquitination, nitration, or SUMOylation, are molecular factors that impact α-synuclein folding, stability, aggregation and, possibly, neurotoxicity [4]. However, recent studies of the molecular architecture of Lewy bodies (LBs), the pathognomonic feature in Lewy body diseases (LBD), are providing new insight into these structures by detecting different proteins and cellular components, in addition to α-synuclein [5], suggesting that α-synuclein aggregation is only part of the process of LB formation. The many potential mechanisms by which α-synuclein misfolding may lead to malignant aggregation are thoroughly reviewed in excellent articles (for detailed reviews, see [6–8], for instance). Here, we will focus on recent reports of a potential prominent role for the receptor for advanced glycation end products (RAGE) in the context of PD, as well as the potential changes that glycation may exert on α-synuclein function, thereby bringing up new insight into α-synuclein pathology and on therapeutic opportunities that may also be relevant for other synucleinopahties.
SYNUCLEINOPATHIES AND α-SYNUCLEIN
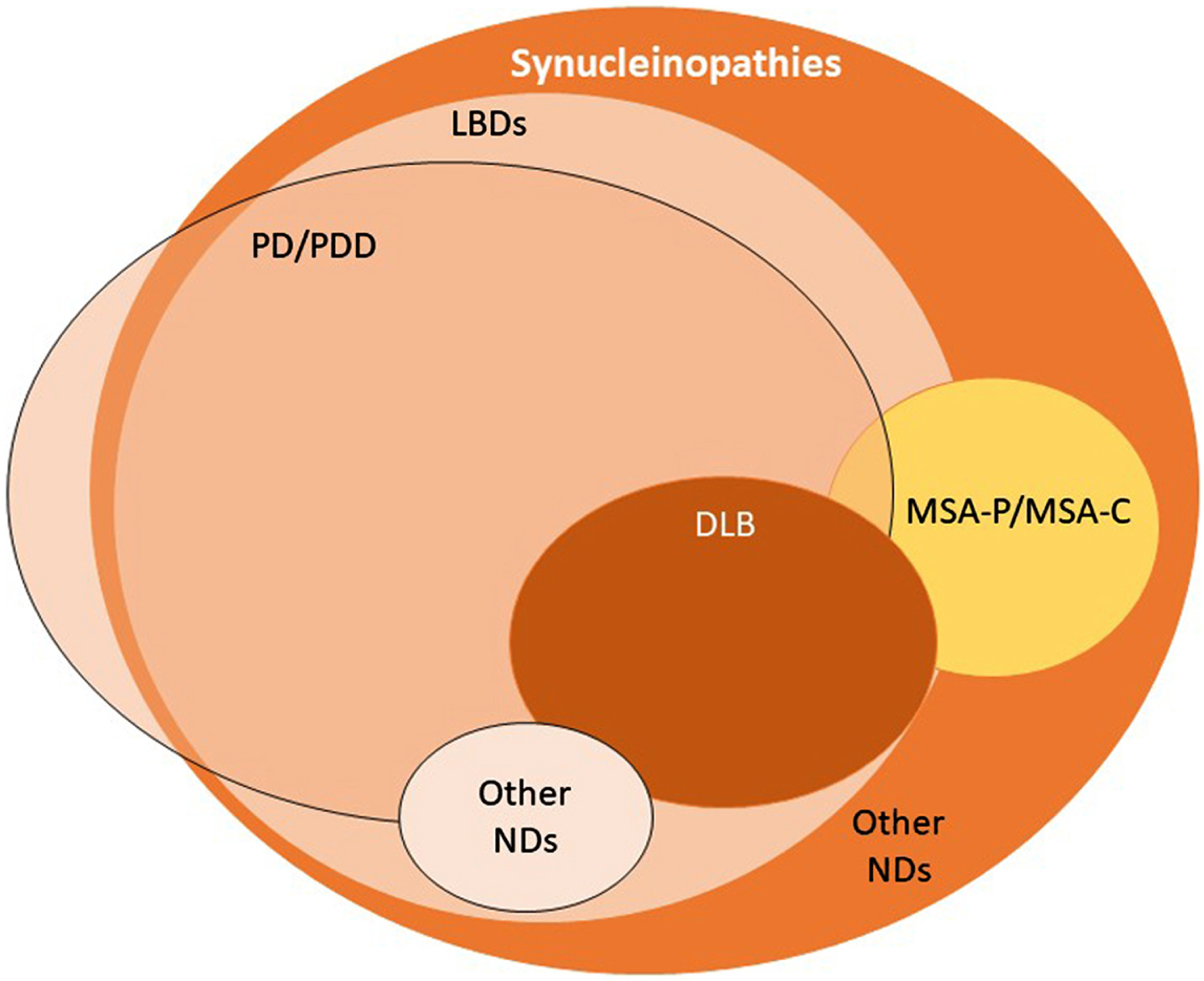
Synucleinopathies are commonly divided into two major groups: LBDs, as mentioned above, and multiple system atrophy (MSA). In LBDs, aggregated forms of α-synuclein are found in the soma and in neurites of neurons (Lewy bodies and Lewy neurites, respectively) [9]. In MSA, α-synuclein inclusions are known as Papp-Lantos bodies, or glial cytoplasmic inclusions, as they are observed in oligodendrocytes [10, 11]. LBDs may manifest as one of a wide group of sporadic or familial pathologies that includes Parkinson’s disease (PD), PD dementia, dementia with Lewy bodies, infantile neuroaxonal dystrophy, autosomal recessive early-onset parkinsonism, adult-onset dystonia-parkinsonism, atypical neuroaxonal dystrophy, Niemann-Pick type C1, Krabbe disease and POLG-associated neurodegeneration (Fig. 1) [12, 13]. Two main clinical subtypes are described for MSA, namely MSA with predominant cerebellar ataxia and MSA with predominant parkinsonism [14]. Synucleinopathies are a relatively common cause of dementia, as PD dementia and dementia with Lewy bodies— collectively referred to as Lewy body dementias (LBD)— account for up to 30% of dementia [15].
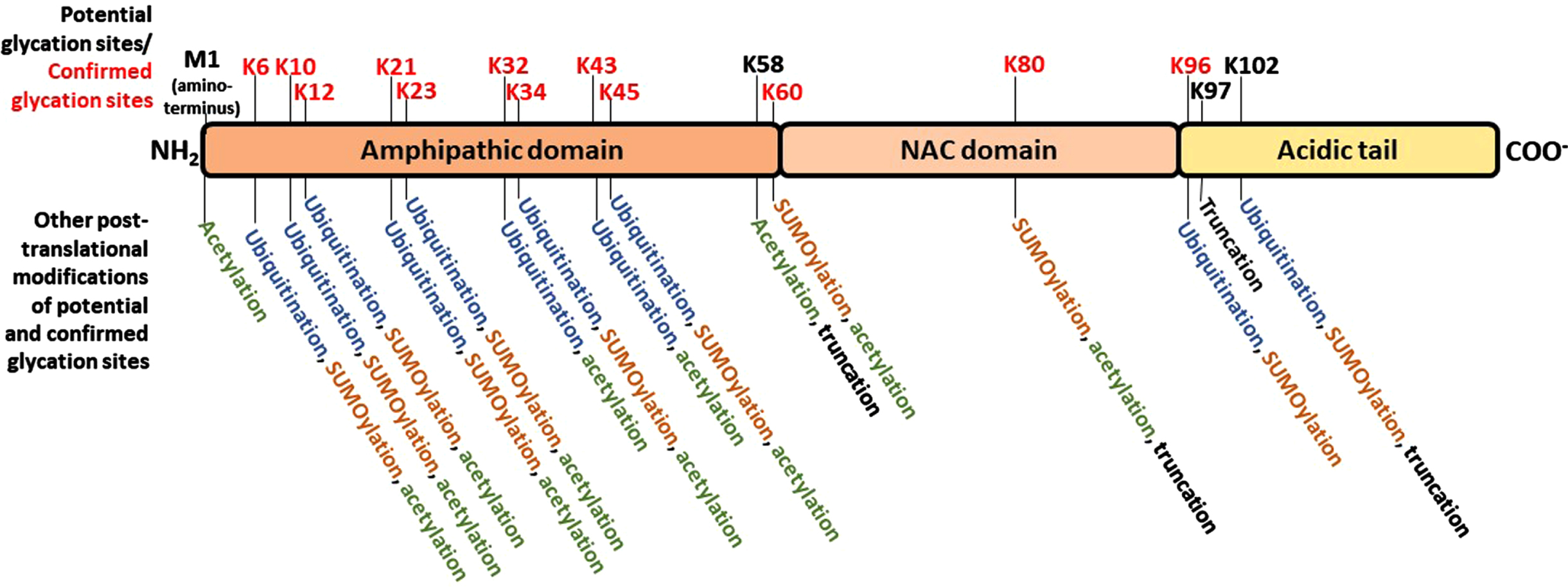
It has been generally accepted that α-synuclein misfolding and aberrant aggregation might be a major driver of genesis and progression of synucleinopathies. α-synuclein is a 14 kDa protein with 140 amino acids encoded by the SNCA gene in humans. It has an N-terminal amphipathic region (1-60 residues) followed by a central portion bearing a highly amyloidogenic domain commonly known as the non-amyloid-β component (NAC, 61-95 residues), and an acidic C-terminal domain (96-140 residues) that is mostly responsible for its classification as an intrinsically disordered protein (IDP) (Fig. 2) [16]. Both the NAC component and truncations of the C-terminal domain play critical roles in α-synuclein aggregation [17, 18]. Due to its localization in pre-synaptic terminals, α-synuclein has long been postulated to exert functions related to neurotransmitter release, synaptic function, and synaptic plasticity [19, 20]. Nonetheless, α-synuclein is expressed also in colon, erythrocytes, peripheral nerves, skin, the urogenital system, and cells of the immune system, including circulating dendritic cells, monocytes, and granulocytes [21]. In pre-synaptic terminals, α-synuclein participates in the fusion of lipid vesicles and induction of membrane curvature [4, 22]. However, α-synuclein can also occur, albeit in lower amounts, in the nucleus, being associated with the regulation of retinoic acid-induced gene transcription [23], and downregulation of cell cycle-related genes. The precise mechanisms regulating the import of α-synuclein into, or export from the nucleus are still unclear, but phosphorylation on serine 129 has been shown to play a role in this process [24].
Despite the described roles in synaptic function, α-synuclein knock out mice do not show major differences in neurological processes, when compared to wild type animals. Intriguingly, they present a deficient immune defense against infectious agents [25, 26]. These observations are connected to the recent demonstration that α-synuclein is essential for the development of a normal inflammatory response to the intraperitoneal injection of bacterial peptidoglycan and for antigen-specific and T cell responses, which led to recent advocations that the main physiological function of this protein would be more related to the immune function than neuronal activity regulation [27, 28]. In fact, several studies demonstrating an important interplay with immune response reinforce this hypothesis. Increased α-synuclein levels has been observed in response to pathogen infections, such as viruses and bacteria, with consequent changes in the function of cells from the immune system [21]. Besides, α-synuclein expression has been observed in the gastrointestinal tissue, and its levels were demonstrated to be modulated in inflammatory bowel disease [29]. Oligomeric α-synuclein interacts with Toll-like receptor (TRL) 4 in macrophages to induce cytokine release, eliciting an immune response against α-synuclein oligomers and possibly an initiating role in PD pathogenesis [30]. Moreover, monocytes release the scavenger receptor for haptoglobin– hemoglobin complexes CD163 in response to activation by either monomeric or fibrillar α-synuclein, and the levels of soluble CD163 in the cerebrospinal fluid and serum of PD patients correlates with α-synuclein levels [31]. Other studies reported that α-synuclein overexpression strongly reduced microglial phagocytic activity and cytokine release [32], while exposure of macrophages to exogenous α-synuclein leads to macrophage polarization and pro-inflammatory activation, inducing TNF-α and IL-1β release through interaction with TLR4 and TLR2 [27, 30]. In microglia, several studies reported the activation of immune pathways in response to α-synuclein, including expression of IL-1β, TNF-α, and IL-6 [21], activation of the NRLP3 inflammasome [33] and immune response mediated by MHCII [34]. These evidences connecting immune responses to α-synuclein have led some authors to propose the classification of the protein as an “alarmin” [27].
GLYCATION OF α-SYNUCLEIN
Glycation is the term applied to the formation of a covalent link between sugars and biomolecules (such as proteins). This process is not biologically controlled, i.e., mediated by enzymes, such as in glycosylation, where a glycosyl donor is attached by an enzyme to a glycosyl acceptor group of a biomolecule in order to form a glycoconjugate. In this context, a glycated protein is totally different from a glycosylated protein. Glycation takes place when sequences of non-enzymatic reactions (e.g., Maillard reactions, Amadori reactions, Schiff base reactions) occur between aldehydes or ketones and amino groups [35]. This means that proteins may be glycated in different parts containing Lys or Arg residues, or in the N-terminal extremity. Since glycation reactions may become irreversible at some point, their products are denominated advanced glycation end products (AGEs). Methylglyoxal (MGO), a byproduct from the glycolytic pathway (and also from other metabolic pathways), is an intermediate of these non-enzymatic reaction sequences and is considered a main causative agent of glycation reactions in biological systems [36]. In fact, mammalian cells express a suitable enzymatic system for MGO detoxification, the glyoxalase system [37, 38].
Glycation affects the aggregation pattern of different proteins involved in neurodegeneration [39]. Interestingly, α-synuclein contains fifteen lysine residues [40], making it prone to glycation at several sites along with the amino terminal group of methionine (M1) (Fig. 2). Theoretically, glycation reactions could take place in any of these sixteen sites, but glycation has been reported in K6, K10, K12, K21, K23, K32, K34, K43, K45, K60, K80, and K96, and not in M1, K58, K97, and K102 [41–44]. However, it is still not clear what are the exact consequences of α-synuclein glycation in the context of PD and other synucleinopathies. What we know is that changes in α-synuclein biology are important in such diseases, and also that glycation is an inevitable process that affects all proteins with aging [45]. Besides that, several studies, including our own, demonstrate that glycation may affect different properties of α-synuclein that may be closely related with neurodegenerative processes of PD, such as malignant aggregation and activation of neurotoxic signaling. Importantly, while this remains to be investigated in MSA and other synucleinopathies, glycated α-synuclein is increased in the neurons of LBD and PD patients [46–48]. We also found that glycation promotes α-synuclein oligomerization, not fibrillization [48]. Although this is still a topic of controversy (as deleterious effects of protein aggregation are often associated with fibril formation), it must be pointed out that the activation of the protein clearance pathways is dependent on the formation of particular types of fibrils; in this context, it was observed that glycation prevented the formation of non-malignant fibrils, while favored the accumulation of more toxic forms of aggregated protein – oligomeric species [49, 50].
As a result of glycation, alterations in functional properties of α-synuclein are also believed to be involved in the molecular mechanisms leading to PD and other synucleinopathies. These include changes in cellular localization, in membrane binding properties, in susceptibility to ubiquitination and, importantly, in reduction in its cleavage in the extracellular space, which may promote intercellular propagation [40, 48]. Ubiquitination of the amino terminal portion of α-synuclein, in particular, is an important process of protein targeting for degradation that may be affected by glycation, since these PTMs compete for many of the same lysine residues (Fig. 2). In vivo, the injection of MGO in transgenic mice expressing human α-synuclein (Thy1 α-synuclein mice), results in the glycation of proteins in the midbrain that are directly associated with dopaminergic pathways involved in PD, and enhances cognitive deficits and motor PD-like phenotypes [51]. However, whether glycation is a causative factor of PD-associated changes in α-synuclein homeostasis, or just another consequence of a more systemic, widespread process leading to neuroinflammation is not known. Studies to investigate the extent of glycation in pathological forms of α-synuclein in LBD and MSA could help to address this issue in the future. In addition, the extent of the susceptibility of α-synuclein to glycation-mediated changes is still unclear. Considering that this protein has a relatively large number of lysine residues (Fig. 2), and the consequences of glycation reported so far, it might be reasonable to expect the existence of anti-glycation or other protein repair mechanisms against glycation in the brain.
In a recent study, we compared the glycating activity of different sugars (MGO, ribose, fructose, mannose, glucose, galactose, sucrose, and lactose) on α-synuclein, and observed that MGO and ribose have the strongest glycation activity, producing aggregates that were very efficient in preventing fibril formation [52]. In most former studies, MGO is the preferred glycating agent used to modify α-synuclein in vitro or in vivo. This inevitably leads us to speculate that α-synuclein glycation in vivo may be associated to conditions in which MGO production or accumulation is increased. Several populational and metadata-based studies suggest a possible relationship between diabetes (mainly type 2) and PD, with observations, for instance, of a 23% increased risk of PD in diabetes patients [53]. However, is this really the right question to ask?
Considering that the exact physiological function of α-synuclein is still unclear, it is not unreasonable to ask if α-synuclein itself may play a role in the homeostasis of glycation and AGE accumulation in the cell. Strikingly, in α-synuclein deficient mice (snca-KO), a significant upregulation in glyoxalase I expression and activity is observed in striatum and cerebellum, and this is accompanied by enhanced accumulation of MGO and AGEs formed from glyoxalase I substrates, indicating that this upregulation is a compensatory response against dicarbonyl stress that takes place in the absence of α-synuclein [54]. Thus, can modulation of brain glucose-related stress be added to the list of the several functions reported for α-synuclein? Although α-synuclein ablation clearly results in deleterious changes associated to glucose-related stress, the mechanism by which α-synuclein expression results in this protection against glycation has never been explored and remains to be addressed in future studies, in order to establish if there is a direct relationship between α-synuclein and glucose metabolism/stress.
RAGE
The receptor for advanced glycation end products (RAGE) is a type-1 transmembrane glycoprotein belonging to the IgG superfamily. It displays one variable (V-type) and two constant (C1-type and C2-type) domains in the extracellular environment. This structure is followed by a transmembrane domain responsible for attachment of cell surface and a cytosolic tail that mediates signal transduction through interaction with diaphanous-1 protein [55–57]. Initially, it was described as a receptor for AGEs, and was implied in the inflammation and tissue damage of diabetes, being considered a key mediator of circulatory complications and nephropathy in this disease [58–60]. Since then, RAGE has been found to recognize several ligands from diverse classes of molecules and with greater affinity than AGEs. These include amphoterin or High-Mobility Group Box-1 protein (HMGB1) [61], S100B and other members of S100 family [62, 63], extracellular nucleic acids [64], amyloid-beta peptide (Aβ) [65], HSP70 [66, 67] and bacterial LPS [68], among other danger- and pathogen-associated molecular patterns (DAMPs/PAMPs). Such ligand behavior included RAGE in the category of Pattern Recognition Receptors (PRRs), which are important elements of the immunological response, such as the toll-like receptor (TLR) family and nucleotide-binding oligomerization-like receptors [69]. This and the wide range of pro-inflammatory pathways activated in response to ligand binding led researchers to assess the role of RAGE in the development of several non-transmissible pathologies where inflammation is a crucial element, such as diabetes, cancer, circulatory/cardiac conditions, and neurodegenerative diseases.
Following the observation that Aβ peptide could bind RAGE, several studies focused on the elucidation of the effects caused by RAGE activation or inhibition in Alzheimer’s disease (AD). A clinical trial has been conducted with a pharmacological RAGE inhibitor, but the results from the PF-04494700 clinical trial did not show conclusive signs of significant improvement in AD patients, even though the drug was considered safe and tolerable [70, 71]. However, as neuroinflammation has been increasingly recognized as a key factor in the onset and the progression of neurodegeneration, observations of the involvement of RAGE in signaling mechanisms that connect inflammation to neurodegeneration have also emerged in other neurodegenerative conditions. The expression of RAGE was observed in experimental models of toxin-induced dopaminergic denervation [72–74], and also in a limited number of people with sporadic PD [75], G2019 S LRRK2 PD [76], and incidental Lewy body disease [46]. Moreover, a study with a Chinese Han population concluded that the RAGE -429T/C polymorphism was associated with increased susceptibility to PD and the CC-genotype of -429T/C could be a protective factor for the development of the disease [77]. Nonetheless, more studies are necessary to evaluate the presence of RAGE in brain areas and cell types affected by synucleinopathies in human subjects, as there is no information currently available for many of the diseases mentioned earlier.
The mechanism by which RAGE is proposed to contribute to pathology is often related to the maintenance of a local pro-inflammatory state in the nervous tissue (Fig. 3). Most known RAGE ligands are DAMPs/PAMPs, some of which also activate other PRRs such as Toll-like receptors 2 and 4 (TLR2/TLR4) [78, 79]. Intracellular signaling triggered by RAGE ligand binding may vary among cells, but the pro-inflammatory effects are mainly ascribed to the activation of Nuclear Factor κB (NFkB)-dependent gene transcription of chemokines and cytokines. Since the RAGE gene (AGER/Ager) also has a responsive element to NF-kB in its promoter region, the presence of RAGE ligands in the extracellular environment is postulated to increase and sustain the local pro-inflammatory state [80, 81]. Additionally, NADPH oxidase activation and consequent enhanced production of reactive species have been reported as another consequence from RAGE-induced signaling, which would further contribute to establish a self-sustaining positive feedback axis of pro-inflammatory activation at tissue level [82, 83]. The observation of the physical interaction between the cytoplasmatic domain of RAGE with TIRAP and MyD88 [78] further reinforces the classical view of a pro-inflammatory modulator contributing to progressive cell death.
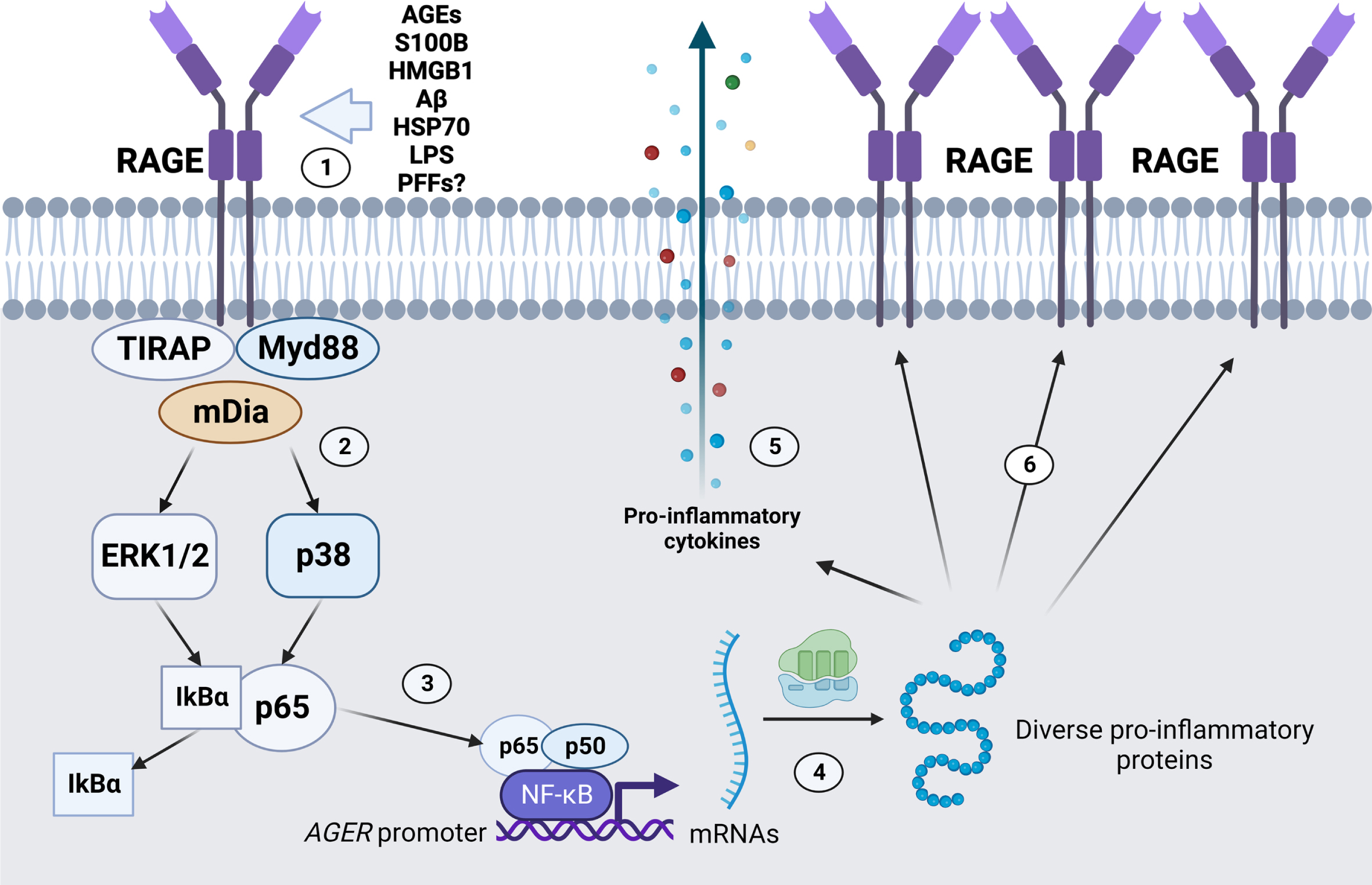
However, as the understanding of RAGE biology progresses, the view that this receptor is solely a contributor to inflammation in neurodegeneration becomes way too simplistic. RAGE is constitutively expressed in lungs, exerting a role that is far from being totally understood and seems to be not primarily related to inflammatory modulation; in fact, disruption of RAGE expression in lungs is associated to lung disease [84]. It was not until 2014 that a phylogenetic study demonstrated that the AGER gene is closely related to homophilic cell adhesion molecules (CAMs) genes (e.g., ALCAM, BCAM and MCAM) [85], which shed light on the relatively neglected cell adhesion properties of RAGE— which, in turn, may explain its constitutive expression in lungs, among other biological activities that will be further discussed here. In this context, a more critical and attentive approach must be applied in order to fully determine the true nature of RAGE and its role in neurodegenerative diseases.
α-SYNUCLEIN AND RAGE: IS GLYCATION NECESSARY FOR THEIR INTERPLAY?
As mentioned earlier, recent observations are consistent with the hypothesis that α-synuclein is a critical mediator of inflammatory and immune responses [26, 87]. In addition to these studies, and the widespread evidence associating inflammation to neurodegeneration in PD, another recent publication has demonstrated that α-synuclein fibrils interact with RAGE, thus adding another protein to the growing list of RAGE agonists [88]. In this study, α-synuclein pre-formed fibrils (PFFs) were found to bind to the V-domain of RAGE with a 50-fold-higher affinity compared to monomeric α-synuclein. According to this study, RAGE is responsible for many of the pro-inflammatory effects by α-synuclein PFFs on microglia, including TNF-α, IL-1β, and IL-6 expression, and p38 MAPK phosphorylation. Besides, monomeric α-synuclein fails to induce the same effects in primary microglial cells and microglial cell lines. Thus, although α-synuclein presents several glycation sites, these results indicate that glycation is not needed for the activation of RAGE. Also, they provide a direct link between pathological α-synuclein and the pro-inflammatory effects by RAGE, indicating that whenever α-synuclein fibrils are formed and RAGE is present, a pro-inflammatory signal cascade is potentially activated, evoking the PD correspondent of the Aβ-RAGE axis observed in AD. Here, however, α-synuclein fibril formation is critical to convert α-synuclein in a pro-inflammatory RAGE ligand.
These data are highly significant to the field, but they also raise new questions. For instance, the role of α-synuclein glycation in the α-synuclein-RAGE interaction must be thoroughly investigated. Since RAGE is capable of recognizing different AGEs and trigger inflammatory responses, are glycated monomers of α-synuclein able to bind and activate RAGE? And what about non-fibrillar aggregated forms of glycated α-synuclein? Questions on the physiological role of this interaction must be pursued in more detail, particularly when considering data provided by in vivo studies that investigate RAGE in the context of PD. This is because the data available so far from studies investigating RAGE in patients and animal models do not indicate that microglial cells is the main site of RAGE expression in PD. In fact, although there are a few studies evaluating the content of RAGE in different parts of the brain and CSF of PD patients by WB and ELISA [75, 76], the identification of the cell type expressing RAGE in human brains affected by PD is still lacking. Nevertheless, studies with different PD animal models that have applied immune-based microscopy approaches to identify the cell types expressing RAGE have observed an almost complete absence of RAGE in microglial cells, as summarized in Table 1. From this compilation of studies that used different toxin-based models (MPTP, 6-hydroxydopamine and rotenone), and one genetic model (G2019 S LRRK2 mutation), it is evident that neurons of the nigrostriatal axis are the most frequent cell type where RAGE expression is observed. This information must still be confirmed in PD patients, as it would imply that the function of the α-synuclein PFF/RAGE interaction in PD could be different from the classical activation/maintenance of the local pro-inflammatory axis by microglia as proposed for AD.
Cellular site of RAGE expression evaluated in PD animal models
Even if confirmed, this information is not enough to indicate that the functions exerted by RAGE are not restricted to the regulation of inflammation in PD. Observations of diverse actions of RAGE at the CNS may provide additional clues to understand the whole range of its biological activities in both physiological or pathological contexts. A system of primary culture of monkey brain capillary endothelial cells coupled with rat pericytes and astrocytes was used to demonstrate that RAGE participated in the transport of oxytocin from the blood capillary to the brain side, and this was confirmed in vivo. This process is mediated through the binding of oxytocin by RAGE on endothelial cells of the blood side and then transport across pericytes to the luminal side in contact with astrocytes, and inflammatory signaling is not involved in this process. Previous studies also described a similar mechanism for transport of Aβ through the blood-brain barrier (BBB). Systemic infusion of labeled Aβ1 - 40 and Aβ1 - 42 resulted in capillary uptake by RAGE-expressing cells in the BBB and accumulation in to the hippocampus and cortex. However, Aβ transport was followed by inflammatory response, as TNF-α, IL-6, and heme oxygenase-1 expression in neurons was activated [90]. The exact contribution of this process for AD pathology is not known, but the fact that RAGE performs a similar function with such different ligands is, at least, suggestive of a physiological role of this receptor in the selective permeability of the BBB, which certainly deserves further examination. Notwithstanding, future investigations on the role of RAGE in the context of PD and other synucleinopathies must consider this potential role of RAGE in the regulation of the transport of certain molecules through the BBB. It is necessary to think beyond the intuitive hypothesis of RAGE serving solely as a pro-inflammatory receptor potentially activated by α-synuclein in the CNS, especially considering the presence of α-synuclein in several organs and cell types outside the brain.
Despite the need for a more robust demonstration of the presence or absence of RAGE in microglial cells in PD (both in animal models and in patients), the observation that the α-synuclein PFF/RAGE interaction induces the production of pro-inflammatory cytokines in cell culture indicates that glycation is not a necessary requisite for α-synuclein evocation of classical RAGE-dependent effects. At a first glance, this may seem contradictory as, originally, RAGE was described as a receptor for AGEs. As pointed earlier, α-synuclein glycation reduces fibril formation, as it favors the formation of oligomeric species. Thus, can we expect that α-synuclein glycation may actually reduce RAGE activation, since α-synuclein preferably binds to RAGE as PFFs, while monomers have low affinity for this receptor? Only detailed studies on the effects of α-synuclein glycation over RAGE binding and activation, as suggested earlier, will clarify this issue, since the main consequence of α-synuclein glycation is the formation of toxic oligomers that are not fibrillar. Moreover, the interaction of RAGE with α-synuclein PFFs was only compared with α-synuclein monomers [88]. In addition, as α-synuclein is expressed in different organs, the possible role of RAGE in the transport of α-synuclein through the BBB should also be explored. Finally, in a hypothetical scenario where RAGE acts on α-synuclein transport through the BBB, it would be important to investigate how this would work when α-synuclein was presented at different degrees of glycation, oligomerization and fibrillization. Maybe various conditions of α-synuclein glycation and aggregation can evoke different responses from RAGE, influencing the degree of activation of pro-inflammatory pathways.
So, can RAGE be part of a system for recognizing and processing α-synuclein in fibrillar or glycated states? Besides, the determination of the effects of RAGE binding by α-synuclein over cell processes other than inflammation (and in cell types other than microglia), and the effects of glycation in the interaction of α-synuclein with RAGE arise as fundamental questions to establish the direction of the research evaluating the role of RAGE in PD and other synucleinopathies.
CONCLUSION
For a better understanding of the role of RAGE in PD and other synucleinopathies, additional studies in cell types expressing this receptor in the brain of patients and reliable animal models are needed. Besides, it is essential that further studies on the interaction between different forms of α-synuclein (i.e., monomeric vs. aggregated, glycated vs. non-glycated, etc.) and RAGE explore different physiological possibilities resulting from this interaction, instead of only focusing on the classical view of RAGE acting in the neuroinflammation positive feedback axis.
Footnotes
ACKNOWLEDGMENTS
DPG is supported by CAPES/Alexander von Humboldt Foundation (Brazil/Germany) #88881.512990/2020-01, Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq, Brazil) #408435/2018-6 and #301175/2019-5, Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS, Brazil) #21/2551-0000073-2 and #16/2551-0000499-4. TFO is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2067/1-390729940, and by SFB1286 (B8).
CONFLICT OF INTEREST
The authors have no conflict of interests to declare. TO is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.