
Abstract
It is widely believed that environmental exposures contribute to the vast majority of late-onset sporadic Parkinson’s disease (PD), alone or via interactions with genetic factors. The search for environmental causes of PD has however been hampered by lack of understanding the prodromal phase of PD development and the difficulties in exposure assessment during this prolonged period. On the other hand, the existence of this prodromal period, along with an increasingly better understanding of PD prodromal symptoms, provides an exciting opportunity to identify environmental factors that initiate PD pathogenesis and/or modify its progression. For prevention efforts, this prodromal stage is of a major interest. Targeting factors that enter the body via the nose or gut has become even more important since the discovery of α-synuclein aggregates in the enteric and olfactory nervous systems. In this paper, we speculate about novel research hypotheses and approaches that may help us better define the role of environment in PD etiology, especially during its extended and complex prodromal phase.
INTRODUCTION
The causes for late-onset sporadic Parkinson’s disease (PD) remain elusive, and PD is likely the cumulative result of numerous genetic and environmental insults and their interactions in the context of brain aging. Research on the environmental triggers and modifiers for PD development is incredibly important for a number of reasons. First, late-onset sporadic PD takes decades to develop, and by the time of diagnosis, neurodegenerative changes are too advanced to decelerate, stop, or reverse. Therefore, our battle against PD critically depends on disease early identification and intervention which in turn rely on a good understanding of disease etiology and actions upon modifiable risk factors. Second, despite recent great success in unveiling the genetic basis of late-onset sporadic PD, genetic findings may only explain a small portion of the cases and cannot be easily extended to disease prevention. On the other hand, during the decades of prodromal stage PD, many environmental factors may come into play at various time points that may trigger PD pathogenesis and modify its progression [1]. It is reasonable to assume that in a majority, if not all, late-onset PD cases, there are environmental contributions that determine or modify the risk and age of PD clinical onset. Unfortunately, we are far away from identifying these factors, defining their roles, and quantifying their contributions. Third, unlike genetic factors, environmental factors are potentially modifiable, and will therefore have profound implications for PD prevention and treatment. Finally, in the face of the rapid growth of aging populations across the globe, PD emerges as the fastest growing neurological disease in terms of both prevalence and death [2, 3]. Adding to this potential public health crisis, recent evidence, albeit preliminary and inconsistent, suggests that there is an increasing trend in PD incidence over the past a few decades [4], possibly indicating a role for environmental factors. Taken together, there is an urgent need for actions to be taken by funding agencies and PD researchers to identify environmental contributions to PD development.
WHAT DO WE KNOW NOW ABOUT ENVIRONMENTAL FACTORS AND PD?
In the past two decades, scientists have identified over a dozen environmental factors associated with the risk of developing PD, and for a majority, findings are reasonably consistent across studies [1, 5]. Examples include inverse associations with smoking [6, 7], coffee drinking [8, 9], vigorous exercise [10, 11], ibuprofen use [12, 13], and plasma urate [14, 15], as well as positive associations with overall pesticide exposure [16, 17], use of specific pesticides [18, 19], and traumatic brain injury [20, 21]. For most of these associations, plausible biological hypotheses have been proposed. However, causal inference for these epidemiological findings has been very difficult. Apart from limited and often inconsistent experimental data, for most of these epidemiological observations, reverse causation is a viable potential explanation - that PD development prior to clinical diagnosis changes lifestyle and behavior rather than the other way around. Possible exceptions are the use of certain pesticides. For example, epidemiological findings on rotenone and paraquat [18, 19] are supported by strong experimental evidence, so much so that these chemicals are being used to generate rodent models for PD therapeutic research [22]. Even for pesticides, there are many important questions unanswered. Therefore, despite its importance and a reasonable accumulation of literature, our understanding of environmental contributions to PD is still in its infancy.
WHAT ARE THE MAJOR CHALLENGES IN SEARCHING FOR ENVIRONMENTAL CAUSES OF PD?
The fact that late-onset sporadic PD takes decades to develop and the lack of understanding of this prolonged prodromal phase present a major challenge to understand environmental contributions to PD. In this disease development paradigm, causative exposures that initiate PD pathological process may have to occur and be documented decades before disease clinical diagnosis, which is often infeasible in epidemiological studies. Further, once neurodegeneration is initiated, many environmental and genetic factors may come into play to modify PD progression during the decades of prodromal disease development. As a consequence, even the most robust epidemiological findings, including those from longitudinal cohorts, are subject to alternative explanations. For instance, smokers have a robust and substantially lower risk for PD than non-smokers in all types of epidemiologic studies [23]. While a causal interpretation that cigarette smoking reduces PD risk is appealing, alternative hypotheses such as reverse causation and confounding by personality and other unknown risk factors are equally possible [24]. Similar analogies can be easily extended to most, if not all, of the presumed “protective” modifiable risk factors afore-mentioned.
Another major challenge is the lack of a good understanding of “environment” which encompasses a broad range of exposures that humans are in contact with, ranging from chemicals (e.g., pesticides), physical agents, microbes and viruses, to climate, lifestyle, socioeconomic conditions, and host-environment interactions. Unlike for the human genome, we do not have a “blueprint” of our environment; adding to the complexity, environmental exposures change over time, and their health consequences are likely to be complex, cumulative, interactive, and dynamic. While these are common challenges in environmental research for almost all chronic diseases, they are particularly challenging for neurodegenerative diseases such as PD. Neurons are long-lived, and thus the same aging neurons are subject to potential environmental influences over our entire lifespan. Further, PD is no longer considered a disease of the brain, it involves multiple systems and organs; when it comes to study environmental contributions to PD, we have to be mindful about environmental exposures through multiple routes of entry and at multiple susceptibility windows, which are yet to be better defined. All this, coupled with a decades-long disease initiation and prodromal development period, makes reliable and valid exposure assessment in epidemiologic studies that captures the most relevant etiological periods very difficult to achieve.
NOW IS A MAJOR OPPORTUNITY TO MOVE FORWARD!
Although still somewhat controversial, the Braak hypothesis [25] presents a unique hypothetical framework of PD development that may allow us to better conceptualize steps of PD prodromal development and environmental contributions. According to this hypothesis, PD Lewy pathology develops in six sequential stages, first in the olfactory bulb or enteric nerves (stage 1), years if not decades, before spreading to the substantia nigra where dopaminergic neuron death occurs (stage 3). In support of this hypothesis, recent clinical and epidemiological studies have clearly documented a wide range of nonmotor symptoms in PD patients, and some symptoms such as olfactory impairment [26, 27], REM sleep behavior disorder (RBD) [28, 29], and constipation [30, 31] may have developed years, if not decades, prior to PD clinical diagnosis. While there are still substantial challenges to adequately define prodromal PD, by using these symptoms as noninvasive intermediate phenotypes, we may be able to bring new insights into this “black-box” of PD prodromal development by identifying factors that initiate PD pathogenesis, lead to these intermediate phenotypes, or modify progression to clinical PD (Fig. 1). This framework may fundamentally improve understanding of PD prodromal development and contributions from environmental factors.
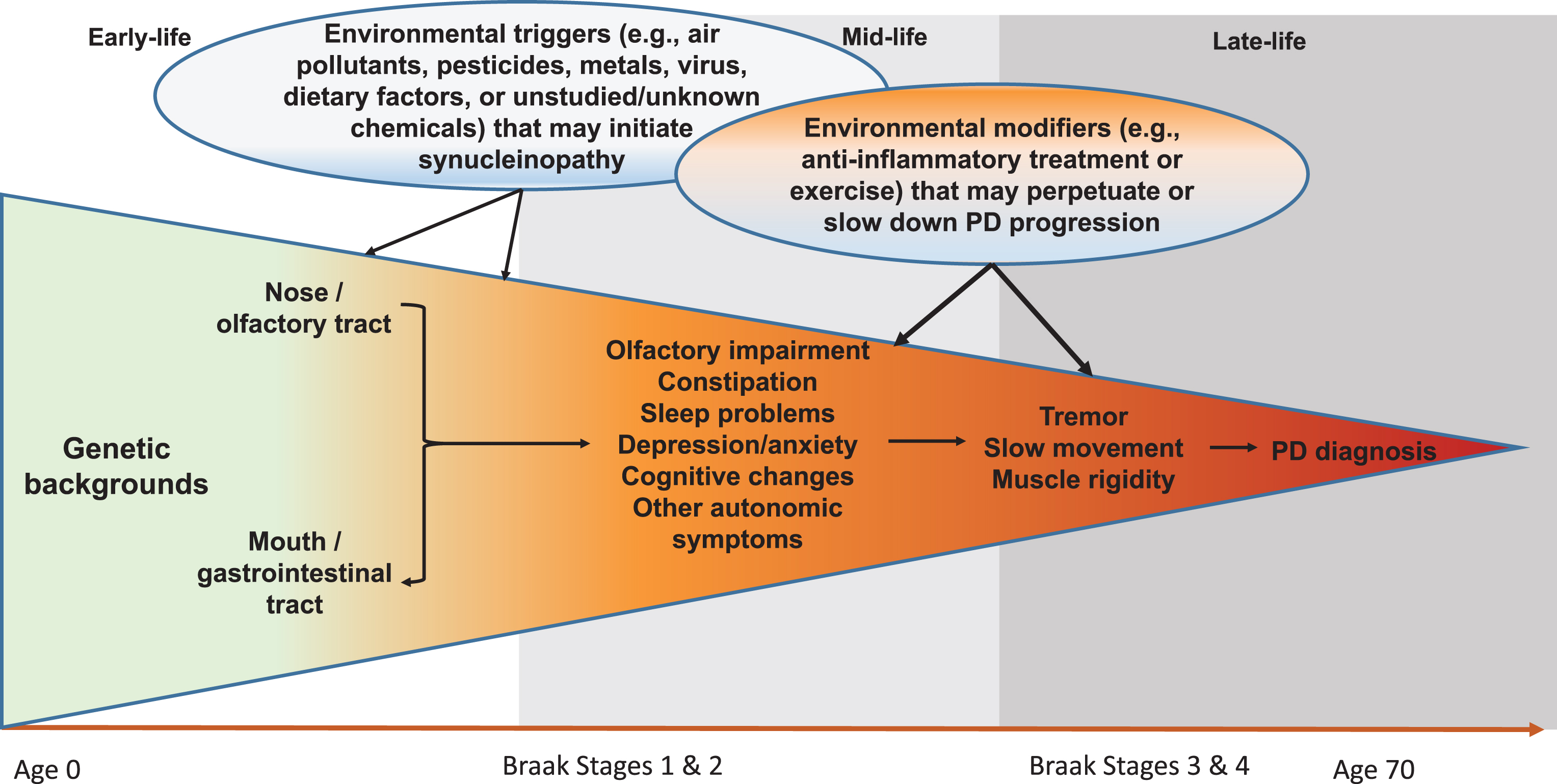
Systematic and Life-long Approach to Study Environmental Triggers and Modifiers for PD Development. Late-onset sporadic PD takes decades to develop. In early- to mid-life, some environmental toxicants (e.g., pesticides, air pollutants, virus) may enter the body via the nose or the mouth, which may induce pathological synucleinopathy in susceptible individuals via mechanisms such as inflammation or microbiome dysbiosis; over time, the pathology may progress to the central olfactory structures and/or the lower brain stem and induce symptoms such as olfactory impairment and sleep disturbances. A portion of affected individuals may further develop motor deficits over years, which may eventually lead to a PD diagnosis. During this prolonged process, many environmental factors may come into play at various time windows to perpetuate or slow down the disease prodromal progression. Modified from Chen H (2018). J Parkinsons Dis.
NOVEL ENVIRONMENTAL PERSPECTIVES
The Braak hypothesis implicates the olfactory pathway and digestive tract as potential origins of PD development [25]. Importantly, these are the two anatomic sites where the human body directly interacts with the environment, where inflammation commonly occurs, and where paths to the brain are well established. It is possible that environmental toxicants such as air pollutants, pesticides, dietary contaminants, or viruses may enter the human body via the nose and/or mouth, and may initiate PD pathogenesis at the olfactory bulb and/or the gut enteric nerves; over time, the pathogenesis may spread to the brain via the olfactory and/or the vagus nerve, and eventually lead to dopaminergic neuron deaths in the substantia nigra [32]. All these observations make investigations of environmental exposures that enter via the olfactory or digestive tracts relevant and attractive for PD etiology. A few common and relevant exposures of interest are discussed below.
Air pollutants
The olfactory pathway represents an entry point for airborne pollutants that bypasses the blood-brain barrier (BBB), and has been shown as a route for pathogenic α-synuclein transmission to the brain [33]. Therefore, it is plausible to hypothesize that airborne pollutants may contribute to PD pathogenesis by initiating α-synuclein neuropathology in the olfactory system, which may later progress to PD in the context of brain aging [33]. Epidemiological evidence on air pollutants and PD is provocative, but limited and somewhat inconsistent [34–37]. Interestingly, α-synuclein aggregates were observed in the olfactory bulb of toddlers, children, teens, and young adults who prematurely died in Mexico City where air pollution is high [38]. In the US where the air pollutant levels are substantially lower, air pollutants have recently been linked to olfactory impairment among older adults [39, 40]. This is accompanied by a growing literature on potential harmful effects of air pollutants on cognitive function and dementia [41], as well as on neuroinflammation as a potential pathway between air pollutants and neurodegenerative outcomes [42]. We expect future studies not only connect air pollutants, olfactory impairment, and PD development, but also ascertain underlying biological mechanisms.
The gut microbiome
The gut microbiome has quickly been recognized as a major contributor to human physiology as it influences the immune system, and it is responsible for the uptake of nutrients, medications, and environmental toxicants. Evidence is mounting that the microbiome can affect various aspects of neurological functions, brain activities, and behaviors in both animal models and human studies [43]. The Braak hypothesis further puts the microbiome to the forefront of PD etiological research [43, 44] as it implicates gut enteric nerves as an initiation site of PD pathology [25]. In support of this, constipation is one of the most prevalent prodromal symptoms of PD [45] which may have developed 1-2 decades prior to PD diagnosis [30, 46]. Recent studies have compared the gut microbiomes of PD patients with those of controls [47–53]. Although results were not entirely consistent, the studies generally identified a pro-inflammatory microbiome in PD patients as compared with controls. Interestingly, one recent study further reported similarities in microbiome dysbiosis between PD and idiopathic RBD patients, suggesting that the microbiome may change in prodromal PD [53]. As the gut microbiome can be affected by many PD-relevant environmental factors (e.g., smoking, dietary factors, and pesticides), examinations of potential environmental influences on the gut microbiome in the context of PD development are warranted. Similar analogies could also be readily made to the nasal or oral microbiomes, which have been largely ignored thus far in PD research [53, 54].
New insights into pesticides and PD development
Pesticides are among the few well-documented harmful environmental contributors to PD [18, 19]. However, when, where, and how specific pesticides contribute to PD development remains largely unknown. Pesticides access the body via inhalation, eating/drinking, or skin contact [55]; therefore, the olfactory or digestive entry points can be readily extended to pesticide exposures. If pesticides enter the body via nose or the digestive tract, they may initiate synucleinopathy in the olfactory structure or gut, which may later spread to the brain over time via olfactory nerve or the gut-to-brain axis [56]. Research on pesticides in relation to constipation and olfactory impairment in the context of aging may provide information critically relevant to PD development. Although olfactory and gastrointestinal symptoms have been documented in pesticide-based animal models of PD [57, 58], human empirical evidence is sparse and indirect. One study in North Carolina found that farmworkers had higher olfactory thresholds than non-farming laborers [59], but the study could not directly attribute this observation to pesticide use. Interestingly, recently evidence suggests that some pesticides alter the gut microbiome. In murine models, the organophosphate insecticide diazinon perturbed the community structure, functional metagenome, and metabolic profile of the gut microbiome [60, 61] by modulation of quorum sensing, a key mechanism that regulates bacterial populations, composition, and importantly, their functional genes. We expect future detailed research to improve our understanding of the roles of specific pesticides in PD development by delineating potential routes of entry, effects at various stages of PD development, and potential biological or pathological mechanisms.
Other relevant environmental exposures
Although not the focus of this article, the Braak hypothesis also provides strong rationales to systematically examine several other environmental exposures that have not been well studied in the context of PD development such as organic solvents [62], high temperature cooked meats and heterocyclic amines [63], respiratory or GI infections and inflammation [64–66], and the use of antibiotics and antiviral therapies [67].
OTHER IMPORTANT CONSIDERATIONS IN DEFINING ENVIRONMENTAL CONTRIBUTIONS TO PD
Gene-environment interactions and epigenetics
Many biologic processes have evolved to be accomplished through multiple mechanisms that involve both genetic and environmental contributions, and both have to come together before a biologic system fails. Identification of genetic factors that modify the association of specific environmental stressors with PD will improve our understanding of etiological mechanisms. The success in understanding PD gene-environment interactions thus far has been largely limited to interactions of genetic factors with specific pesticides. For example, studies suggest that the potential adverse effects of paraquat on PD may be substantially augmented by genetic variants that are related to dopamine transporter function [68] or DNA base excision repair failure [69], possibly by rendering the system unable to compensate for the combined synergistic insults. While these individual studies provide crucial leads in understanding specific gene-environment interactions and pathways, we expect increasingly available and more statistically powerful pooled analyses to identify [70, 71], cross-validate, or refute [72] potential gene-environmental interactions in PD etiology.
Epigenetic research is another exciting approach to understanding PD pathogenesis by studying mechanisms that regulate gene function through DNA methylation and histone modification without alternating DNA sequence itself. Most PD epigenetic studies to date have targeted methylation in candidate genes using blood or saliva samples [73], and interpretation of study findings are limited by the fact that epigenetic regulations are often tissue specific. Nevertheless, recent epigenome-wide association and network analyses showed that PD status was associated with DNA methylation changes in blood and saliva that implicated the immune system, with some of the differences not due to differential white blood cell composition that distinguished PD patients from controls [74, 75]. This observation converges nicely with the increasing genomic [76, 77], epidemiologic [66], and experimental [78] evidence for the importance of immunity and inflammation in PD development. Epigenetic approaches, as well as other ‘omics’ tools (proteinomics, transcriptomics, lipidomics, metabolomics), based on human tissue samples and induced human stem cells can also be employed for the “meet in the middle” approach, an informative strategy for the integration of such technologies into epidemiological research [79]. We expect such synergistic approaches to provide more insights into biological plausibility and to help establish causality of epidemiological findings.
A life-long and exposome approach to PD etiological research
Most epidemiological studies on PD have focused on a snapshot of a single environmental exposure often in mid to late adulthood, largely ignoring the multidimensional complexity of the disease that may require a life-course perspective. Some experimental studies showed that prenatal or early developmental exposures to certain pesticides (e.g., dieldrin [80], paraquat or maneb [81], atrazine [82]) can lead to dopaminergic neuron degeneration later in life. Others reported that such early-life exposures rendered animals more susceptible to a second toxic environmental insult later in life [81, 83]. These findings suggest that early-life exposures may be contributing to PD development by setting the stage for late-life susceptibility to PD, and point out the importance of preventing repeated exposures during vulnerable periods. Although a life-course approach to late-onset neurodegenerative diseases such as PD is methodologically formidable, we expect future studies to carefully consider such possibilities.
The latest genetic research embarked on generating a polygenic risk score that encompasses all known risk alleles and its use has been promoted to define individual’s overall genetic susceptibility to PD [84] or to predict disease progression [85]. In comparison, a “composite environmental index” is much more difficult to develop, and a vast majority of the epidemiological studies have focused on a single risk factor (e.g., smoking). Nevertheless, epidemiological studies have made first efforts to simultaneously consider multiple environmental exposures. For example, Lee et al. reported potential joint effects from traumatic brain injury and paraquat exposure on PD risk [86] and Kim et al. simultaneously considered smoking, caffeine intake, physical activity and several other factors [87]. These findings are provocative, suggesting stronger associations when composite exposures and their possible synergism are considered. In addition, biomarkers of long-term and lifelong environmental exposures are being developed, such as blood methylation profiles for long-term organophosphate exposures [88]. Further, some major scientific and technological advances that inform the external assessment of the exposome have been made [91, 92], including geographic information systems, remote sensing, global positioning system and geolocation technologies, portable and personal sensing, including smartphone-based sensors and self-reported questionnaire assessments relying on Internet-based platforms. However, all of these methodological and technological improvements in exposure assessment need to be accompanied by new data analysis and interpretation methodologies and also require developing protocols for ethical sharing of sensitive data.
When it comes to considering multiple exposures over time, the concepts of “exposome” and “neuroexposome” are particularly appealing. Chris Wild first proposed the concept of “exposome” more than a decade ago as a measure of the totality of human environmental exposures over one’s lifetime [89]. Heffernan and Hare [90] recently further adapted this concept for neurological diseases. They proposed to cultivate the strengths of “omics” technologies (e.g., genomics and metabolomics), traditional epidemiological surveys, and detailed clinical assessments across the lifespan while considering the unique features of the brain (e.g., BBB permeability and neuron longevity).
This exposome concept, while challenging to implement in the real world, together with the Braak hypothesis, provides a theoretical framework for scientists to design future studies to decipher the environmental causes of PD and develop early interventions to halt the progression to the characteristic motor dysfunction in PD.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
Dr. Chen is supported by a start-up fund from Michigan State University (GE100455), the Parkinson’s Foundation (Grant No. PF-IMP-1825), and the Office of the Assistant Secretary of Defense for Health Affairs, through the Parkinson’s Research Program (Award No. W81XWH-17-1-0536). Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. Dr Ritz was supported by the NIEHS grants ES10544, P01ES016732, U54ES12078, R01ES013717, R21ES022391, R21ES024356, and the NINDS grant: P50NS038367 and by pilot funding from SCEHSC# 5P30 ES07048 and The American Parkinson Disease Association. We would also like to thank Mr. Frank Purdy for proof reading the manuscript.