
Abstract
Background:
Late onset Pompe disease (LOPD) is rare and generally manifests predominantly as progressive limb girdle muscle weakness. It is linked to the pathogenic mutations in GAA gene, which leads to glycogen accumulation in various tissues.
Materials and methods:
We describe the unusual clinical, biochemical, histopathological and genetic characteristics of 5 cases of LOPD.
Results:
The first case had progressive anterior horn cell like disease (AHCD) that evolved later to classical limb girdle syndrome and respiratory failure, the second patient had rigid spine syndrome with gastrointestinal manifestations, the third had limb girdle weakness superimposed with episodic prolonged worsening and respiratory failure, the fourth had large fibre sensory neuropathy without primary muscle involvement and the fifth presented with classical limb girdle muscle weakness. Two homozygous missense mutations c.1461C > A (p.Phe487Leu) and c.1082C > T (p.Pro361Leu) in the GAA gene were identified in case 1 and 2 respectively. Case 3 was compound heterozygous with inframe c.1935_1940del (p.Val646_Cys647del) and an intronic splice effecting variant c.-32-13T > G. Compound heterozygous missense variants c.971C > T (p.Pro324Leu) and c.794G > A (p.Ser265Asn) were identified in case 4. Case 5 had a frameshift insertion c.1396dupG (p.Val466GlyfsTer40) and a synonymous splice affecting variant c.546G > T(p.Thr182=).
Conclusion:
We are describing for the first time from India on LOPD with unusual phenotypes identified. A high degree of clinical suspicion and diagnosing rare phenotypes of Pompe disease is imperative to consider early initiation of Enzyme Replacement Therapy (ERT).
INTRODUCTION
Pompe disease (Glycogen Storage Disease (GSD) type II, OMIM #232300) is a lysosomal glycogen storage disorder (LSD) caused by deficiency of the enzyme α-glucosidase (GAA). The prevalence is estimated to be 1 in 27,800 births [1]. In a recent study, the detection rate of Pompe disease was as high as 1 : 5463 in newborn screening for LSDs [2]. Pompe disease is classified as early (infantile) or late-onset (childhood/juvenile/adult) in accordance with the age of symptom onset and rate of disease progression [3]. It is linked to the pathogenic mutations in the GAA gene, which leads to glycogen accumulation in various tissues. Progressive muscle weakness is known to occur across the disease spectrum. The classic infantile form, characterized by muscle weakness and hypertrophic cardiomyopathy, is invariably fatal within one year of life. Late-onset Pompe disease (LOPD) generally manifests as varied phenotypes. The most common form resembles progressive limb-girdle muscle weakness associated at times with early respiratory muscle involvement [4]. Muscle symptoms are nonspecific, and diagnosing Pompe disease from other neuromuscular disorders is generally tricky. Muscle biopsy showing glycogen accumulation on histology and electron microscopy is a valuable clue to the diagnosis. Estimation of GAA enzyme activity in dried blood spot (DBS) or leucocytes/muscle cells/skin fibroblasts along with identification of recessive mutations in GAA gene clinches the diagnosis and also helps in the phenotypic correlation of infantile vs. late-onset Pompe disease. While complete deficiency of enzyme activity (< 1%) is associated with severe infantile-onset disease, partial deficiency (2–40%) usually correlates with late-onset phenotypes [5].
Generally, biallelic null or truncating mutations are commonly associated with the infantile form with complete loss of enzyme activity, and late-onset phenotypes tend to have variants resulting in partial GAA deficiency like missense and some splice variants [6, 7]. In particular, the c.-32-13T > G intron 1 variant has been reported as the most common mutation in adult-onset GSD II [8].
Here, we describe five unusual phenotypes of genetically confirmed LOPD. The first case presented as progressive anterior horn cell disease (AHCD) that evolved later to classical limb-girdle syndrome and respiratory failure, the second patient presented as rigid spine syndrome with gastrointestinal manifestations, the third with limb-girdle weakness superimposed with prolonged episodic worsening and respiratory failure, the fourth patient presented with large fibre sensory neuropathy without primary muscle involvement and the fifth presented with limb-girdle muscle weakness. Thus, a high degree of clinical suspicion and diagnosing rare phenotypes of Pompe disease is imperative to consider early initiation of Enzyme Replacement Therapy (ERT).
MATERIALS AND METHODS
Institutional ethics committee approval was obtained for collecting the patient data from the medical records of all the patients. The clinical, biochemical, electrophysiological, histopathological, and genetic findings are described in detail for 5 patients with late-onset Pompe disease.
Acid α-glucosidase activity in DBS was measured fluorometrically using a modification of an assay described by Nestor Chamoles et al. [9]. A 3.2 mm spot was punched from each DBS sample and extracted in 270 uL water for 1 hour at 10°C with shaking at 300 rpm. Substrate solution A (1.4 mmol 4-methylumbelliferyl- α-d-glucopyranoside) was prepared by adding 70 mmol/L 4-MUG in DMSO solution to 40 mmol/L sodium acetate (pH- 3.8) and substrate solution B (1.4 mmol 4-methylumbelliferyl- α-d-glucopyranoside) was prepared by adding 70 mmol/L 4-MUG in DMSO solution to 40 mmol/L sodium acetate (pH-7.0). 50 uL of substrate solution A (pH 3.8) and substrate solution B (pH 7.0), 10 uL of 8μmol/L acarbose, and water was added to 30 uL DBS extract in new black 96-well plates. The samples were covered with an aluminum seal and incubated in a shaking water bath at 37°C at 225 RPM for 20 hours. The reaction was terminated by the addition of 150 mmol/L EDTA. A standard curve of 4-Methylumbelliferone (4-MU) (0–2.5–mol/L) was prepared in duplicate by diluting 25–mmol/L of 4-MU in DMSO in water. A quantity of 150–mmol (pH-11.5) EDTA was added to stop the reaction. The plate was read on a fluorimeter at 355 nm excitation and 460 nm emission wavelengths. From the standard curve, molar product quantities were calculated by linear regression. GAA activity of the sample is calculated by subtracting the individual blank activity.
Genetic analysis was done by Next-generation sequencing (NGS) based targeted gene capture using custom-designed clinical exome panel in all five patients using DNA extracted from blood. Sequencing was performed on Illumina platform. Sequence alignment was done to the human reference genome (GRCh37/hg19) and all patients were screened for potential disease-causing variants in known neuromuscular genes curated from online muscle table [10] (GAA variants identified were annotated as per Ensembl transcriptENST00000302262.3. Reported GAA mutations were correlated from Pompe mutation database (http://www.pompevariantdatabase.nl/) for phenotype correlation and functional effect on GAA expression. The variant classification was done based on the American college of medical genetics (ACMG) criteria [11].
Case 4 underwent superficial peroneal nerve, and peroneus brevis muscle biopsy and the muscle tissue was subjected to non-enzyme histochemical staining that included Haematoxylin Eosin (HE) stain, modified Gomori trichrome (MGT) along with periodic stain with and without diastase stain and enzyme histochemical stains included succinic Dehydrogenase (SDH), Nicotinamide adenine dinucleotide Tetrazolium Reductase (NADH -TR), adenosine triphosphatase (ATPase) [pH 9.4, 4.6] and succinic dehydrogenase–cytochrome C oxidase (SDH-COX). Muscle sections were studied under Tecnai G2 Spirit (BioTWIN, Czech Republic) transmission electron microscope (EM). The nerve biopsy was fixed in 3% glutaraldehyde and subjected to routine H & E staining along with connective tissue and myelin stains. The nerve tissue was also subjected to electron microscopic study similar to the muscle.
RESULTS
Case 1
A 20-year-old man born to consanguineous parents was evaluated during 2012 with progressive proximal muscle weakness, recurrent buckling, and tremulousness of fingers for 2 years. There was no muscle wasting, exertion-induced myalgia, or cramps. He had no symptoms to suggest involvement of heart, respiratory or gastrointestinal systems. Examination revealed tongue wasting with fasciculations, prominent bilateral minipolymyoclonus, and mild wasting of shoulder girdles. According to modified Medical Research Council (MRC) grading, the muscle strength was grade 3 at shoulder girdles, grade 2 at hips, and 3+ at the knees. Distal muscles were normal. Tendon reflexes were hyperactive in upper limbs (UL) and exaggerated in lower limbs (LL). Finger flexion and Hoffmann’s reflexes were hyperactive. Babinski’s sign was absent. He had a waddling gait. His serum creatine kinase (CK) level was 3212 IU/L (normal range: 24–229 IU/L), and serum lactate dehydrogenase (LDH) was 736 IU/L (140–248 IU/L). Serum glutamic oxaloacetic transaminase (SGOT) was 173 U/L (8–50 IU/L) and serum glutamate pyruvate transaminase (SGPT) was 148 U/L (7–50 IU/L). Concentric needle electromyography was performed in bilateral biceps brachii, first dorsal interosseous, quadriceps, and tibialis anterior (TA). There was evidence of increased insertional activity, fibrillation, and positive sharp waves in LL muscles. Fasciculations were observed in TA and complex repetitive discharges in quadriceps and TA muscles. Motor unit potentials were normal. TA and biceps showed 75% and 11% polyphasic, respectively. Recruitment was complete in UL’s an incomplete in LL muscles. Motor and sensory nerve conductions were normal. Neuropsychological assessment revealed normal findings. Pure tone audiometry showed normal hearing. Alpha-glucosidase enzyme assay for Pompe disease by Fluorometry method revealed a GAA activity of 0.5 nmol/L/hour (reference range: 19.02–152.22 nmol/L/hour. Biceps muscle revealed polygonal myofibers with prominent large vacuolation in several fibres containing PAS positive and diastase digested large bluish granules suggestive of glycogen accumulation (Fig. 1). Modified Gomori Trichrome staining further highlighted these granules. Acid phosphatase was positive in these vacuoles characteristic of glycogen aggregates (Fig. 1). Cytochrome C Oxidase and other oxidative stains revealed no evidence of mitochondrial disease. These vacuoles were seen in both Types I and IIC fibers. Oil Red ‘O’ stain for lipids was negative. There was no neurogenic atrophy. Pulmonary function tests revealed a forced vital capacity (FVC) of 1.83 litres (39.8% predicted) and forced expiratory volume (FEV1) of 1.78 litres at 1 second (45.2%). The FEV1/FVC ratio was 97.5%. The mid and maximum expiratory flow rates were also low (65%). The inspiratory vital capacity was 1.82 litres (39.6%), and maximum voluntary ventilation was 71.2 litres/min (45.1%), consistent with a severe restrictive pattern.
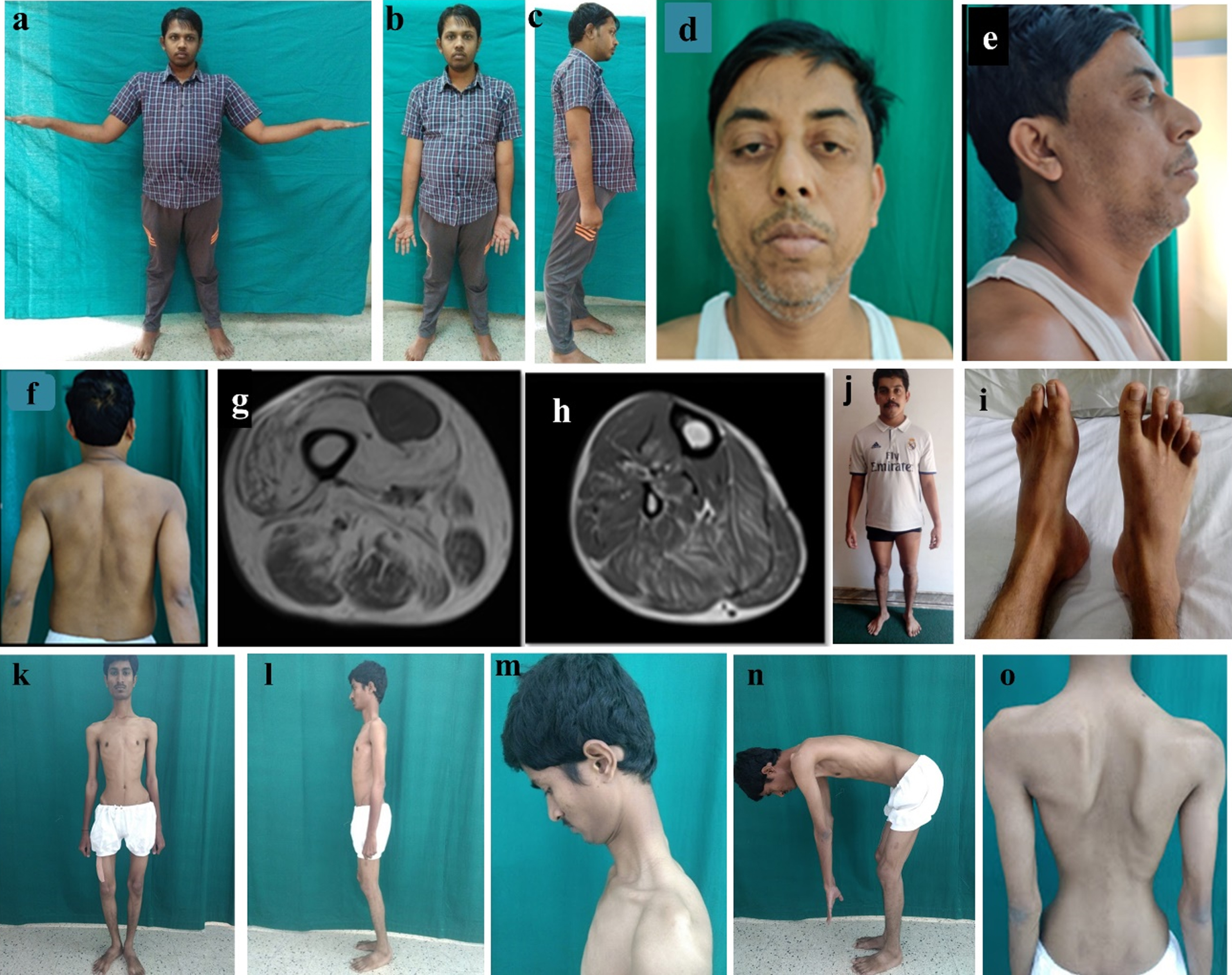
Case 1 (a–c): The proband after 7 years of illness duration demonstrating severe proximal muscle weakness, (b) no distal wasting and (c) protuberant abdomen with mild lumbar lordosis; Case 3 (d, e, f): Note bilateral ptosis, bilateral drooping of angle of mouth suggestive of facial muscle weakness, retrocollis secondary to neck flexor weakness, scapular winging, (g, h) T1 weighted images of muscle MRI showing diffuse fatty infiltration with relative sparing of rectus femoris and tibialis anterior; Case 4. (i) wasting of bilateral tibialis anterior, (j) pes cavus and hammer toes; Case 2 (k–o): thin slender habitus, knee contractures, rigid posture, nuchal rigidity and dorso-lumbar spine rigidity.
Muscle MRI (T1W) showed atrophy and moderate fatty infiltration in quadriceps femoris, gracilis, and hamstring muscles with sparing of the long head of biceps femoris (Fig. 1). In the legs, only TA showed mild fatty changes. Short T1 Inversion Recovery (STIR) sequences showed myoedema in both TA, medial and lateral gastrocnemius muscles (Fig. 1). Brain MRI was normal.
At last follow-up during March 2020, (7.5 years), the patient has profound proximal weakness (Grade 2+/5) with wasting and moderate distal weakness (Fig. 2). He is wheelchair-bound with prominent respiratory distress and severe orthopnea. The tendon reflexes are absent. Repeat cardiac evaluation is normal. Muscle MRI showed further changes with moderate degree fatty infiltration in posterior leg muscles (especially medial gastrocnemius) with myoedema, severe atrophy, and fatty infiltration in quadriceps, adductors and gracilis. The abnormalities in anterior leg muscles were stable. CT of head showed fatty changes in intrinsic tongue muscles (Fig. 3). Ultrasound of abdomen was normal. For the last 3 years he is on enzyme replacement therapy with a stable course.
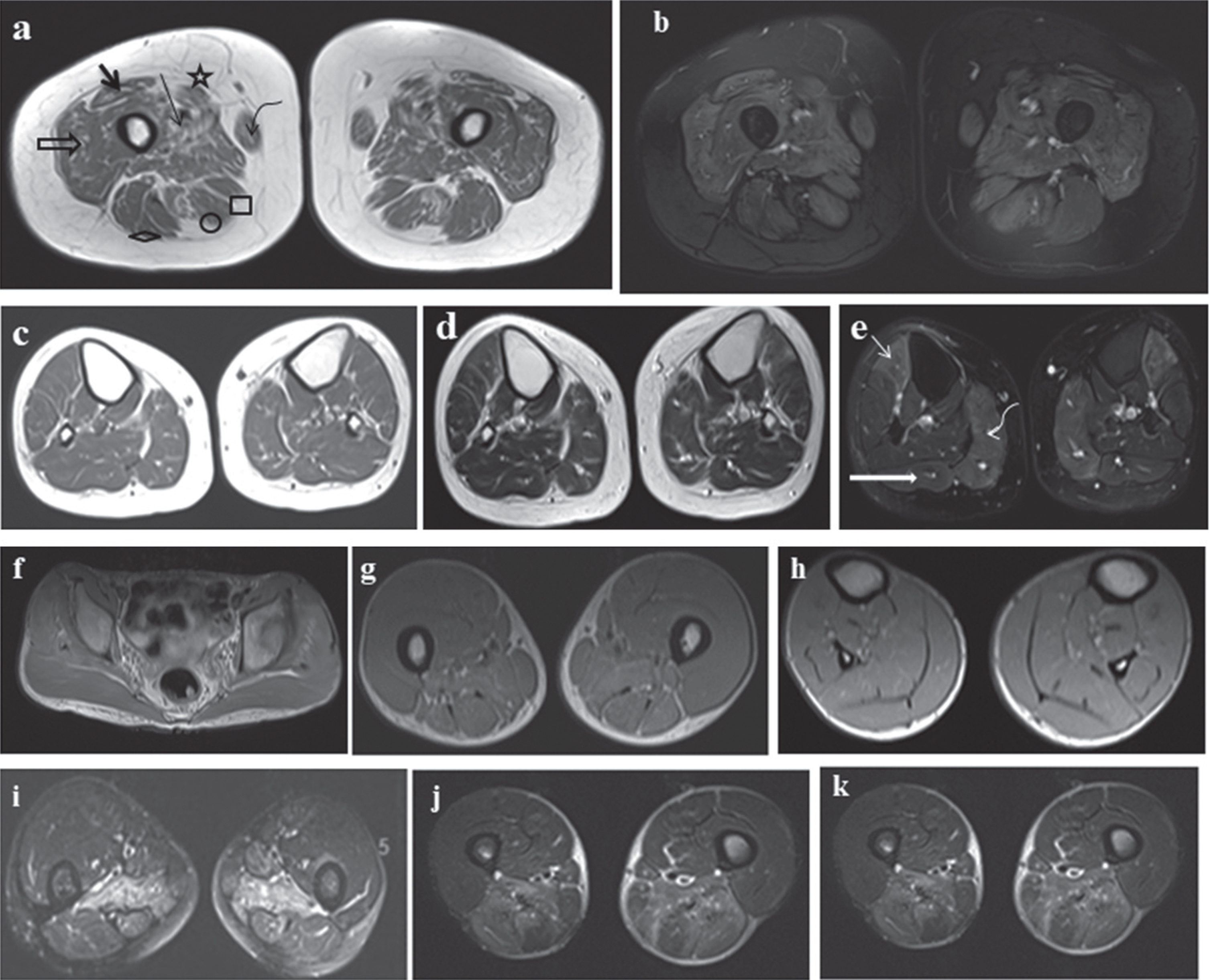
(a–e) MRI of the lower limbs: (a) TI weighted image of thigh showing atrophy with fatty changes in rectus femoris (bold arrow), vastus lateralis (open arrow), vastus medialis (thin arrow), sartorius (star), gracilis (curved arrow), semimembranosus (square), semitendinosus (circle) with relative sparing of biceps femoris (diamond). (b) STIR sequence showing global myoedema of thigh with sparing of right biceps femoris. (c, d) TI and T2 weighted image of Leg: Normal appearance of all muscles except mild fatty changes in bilateral tibialis anterior. (e) STIR sequence: Tibialis anterior (thin arrow), medial head of gastrocnemius (curved arrow) and lateral head of gastrocnemius (bold arrow). These muscles appear hyperintense suggestive of myoedema. (f–h) T1W images showing no fatty changes in hip, thigh and leg muscles. (i) Atrophied paraspinal muscles. (j, k) selective involvement of posterior thigh (adductor magnus and biceps femoris) muscles showing muscle edema on T2 fat suppression images.
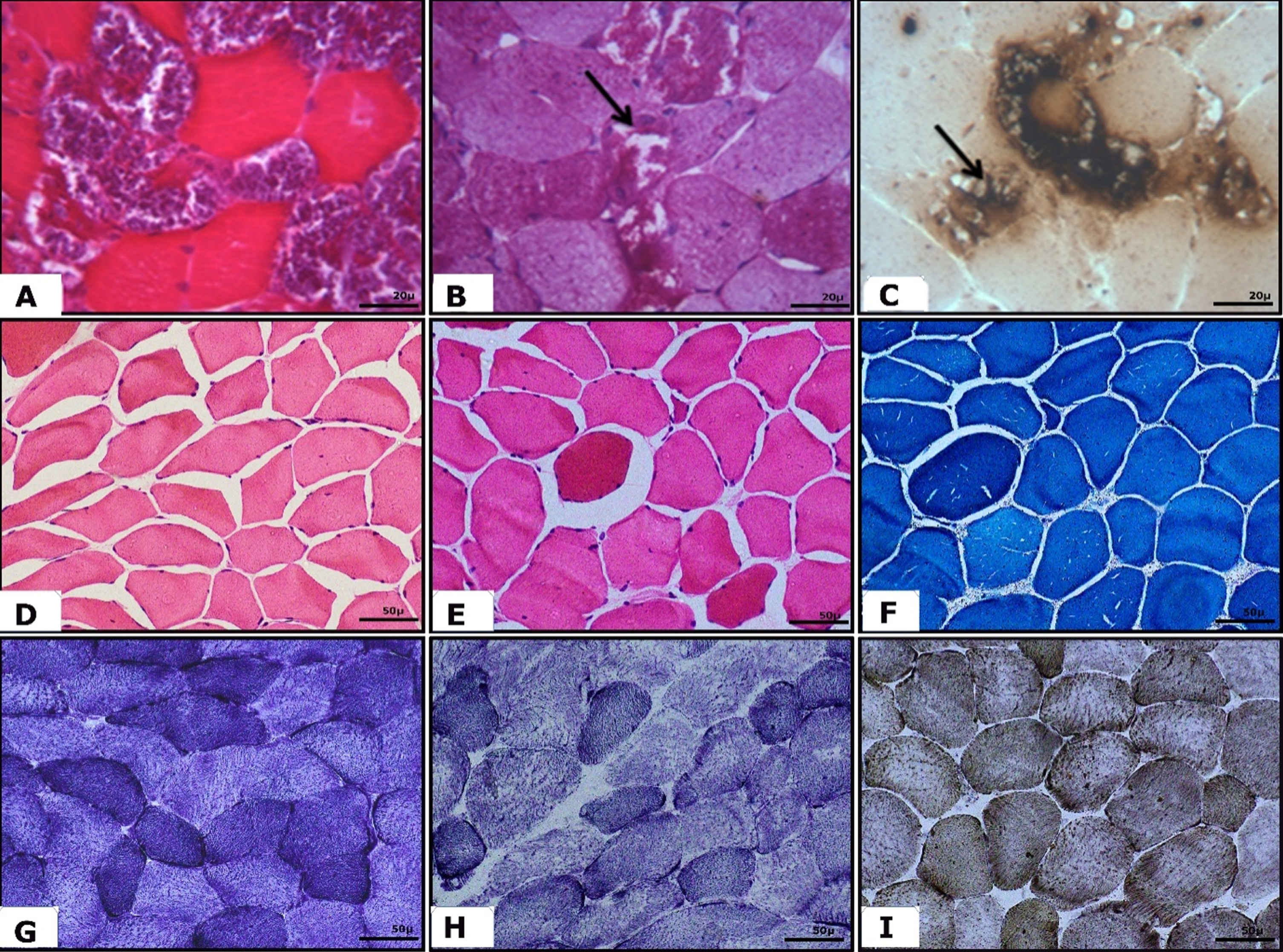
A: Microphotograph showing transverse sections of muscle with variation in fibre size (A – Masson Trichrome stain ×400) along with a few fibres showing sarcoplasmic deposits which are positively stained for periodic acid Schiff (arrow, B – ×400) and acid phosphatase (arrow, C × 400). C, D, E: Microphotograph showing transverse sections of muscle with mild variation in fibre size comprising of a few scattered angulated atrophic fibres with a few internalised nuclei. D, E: H&E ×200; F: MGT -×200; E, F, H: Microphotograph showing Oxidative enzyme staining with no significant histological changes. G – NADH ×200; H - SDH stain ×200; I – Cytochrome oxidase ×200.
Case 2
A 22 year old man from South India born to consanguineous parents presented with recurrent abdominal pain and loose stools from childhood. He had progressive stiffness and restricted movements of neck and spine and slowly progressive limb-girdle weakness from 13 years of age associated with exercise-induced mild fatigue, muscle pains, and occasional respiratory difficulty. He had a lack of appropriate weight gain for age. No cranial muscle involvement. Examination revealed a thin built man with marfanoid habitus. He had prominent nuchal and axial contractures with mild distal hyper-extensibility and thoracolumbar scoliosis with scapular winging (Fig. 4). Muscle strength of upper limb proximal muscles was MRC grade 4, grade 3 at hips, and grade 4 at knees. Distal muscles were normal. Beevor’s sign was positive with a waddling gait. Serum CK level was 962 IU/L. Pulmonary function test revealed a severe restrictive abnormality. Electrocardiography and 2D ECHO were normal. Muscle biopsy was suggestive of subtle myopathic features with no evidence of vacuoles. Muscle MRI of lower limbs showed early involvement with muscle edema of adductor magnus, biceps femoris, and atrophy of paraspinal muscles (Fig. 4). There was no fatty change observed in lower limb muscles.
Dried blood spot (DBS) based GAA activity assay showed low levels (Total α – glucosidase without ascarbose = 4.65 nmol/ml/hour, lysosomal α glucosidase with ascarbose = 0.35 and ration of 0.075).
Case 3
A 50 year old man born to non-consanguineous parentage initially presented to our institute at 30 years of age when SMA was considered. He was lost for follow-up and visited our hospital again in January 2020 with rapidly progressive LGMW with respiratory difficulty for four months. He had a protracted, slowly progressive LGMW from 20 years of age with exertion-induced breathlessness and muscle cramps. From 35 years of age, he has experienced 5 episodes of profound muscle weakness with bed-bound state precipitated by a febrile illness / unusual physical exertion. Each episode lasted from one to 2 months with spontaneous recovery to his premorbid state. On general examination, he had protruded flabby abdomen, paradoxical breathing with a chest expansion of 0.5 cm. Cardia was normal. There was asymmetrical ptosis with mild ophthalmoparesis, mild bifacial weakness, left scapular winging, wasting of arms, shoulder girdles, and quadriceps muscles. Muscle strength was grade 4- at shoulder, 4 at the elbow, 2 at the hip, and 3 at extensors of the knee, and remaining limb muscles were grade 5. Neck flexors, truncal and abdominal muscle weakness were profound. Beevor’s sign was prominent and was evident even during regular breathing. Tendon reflexes were normal. There were no sensory or cerebellar signs. Investigations including complete blood count, liver and renal functions, serum ammonia, and lactate levels were normal. TSH was 11.9 microIU/ml with normal free T4 and T3 levels. He had dyslipidemia. CK level was 370 IU/L. NCS showed normal motor and sensory conductions with no decremental response. No spontaneous activity was noted on EMG. MRI muscle showed diffuse fatty infiltration with relative sparing of rectus femoris and tibialis anterior muscle (Fig. 4).
Left quadriceps muscle biopsy done at 31 years of age revealed mild fiber size variation with occasional hypertrophic fibers, sarcoplasmic degeneration, and focal centronuclear migration. Special stains were not performed. DBS revealed low levels of GAA (total acid alpha-glucosidase level (A) – 19.93 nmol/hr/ml, lysosomal acid alpha-glucosidase(B)- 1.47 nmol/hr/ml and ratio (B/A- 0.07).
Case 4
A 25 year old male, born to non-consanguineous parents with normal developmental milestones presented with inability to grip the footwear and weakness of hand muscles with in advertent dropping of small objects from the hands for 17 years. He had a symptom of washbasin phenomenon. There was no proximal muscle weakness, cranial nerve, or sphincter involvement. No history of muscle cramps or exercise intolerance except for occasional myalgias.
On examination, he had pes cavus, hammertoes, clawing of hands, wasting of foot muscles, and scoliosis (Fig. 5). The muscle tone was normal, and strength was grade 5 in proximal muscles and grade 4 in distal muscles of upper and lower limbs with absent tendon reflexes. Sensory examination revealed the absence of pain and touch sensations up to the wrists and ankles with loss of joint position sense at toes bilaterally. Vibration sense was lost till the anterior superior iliac spine and reduced in both upper limbs at the radial styloid process. Investigation revealed normal liver function tests and CK levels (116 IU/L). Tandem mass spectroscopy was negative for any abnormal metabolites, but serum lactate level was mildly elevated at 25.2 mg/dl. Electrophysiological studies detected absent sensory nerve action potentials (SNAP) with normal compound muscle action potentials (CMAP). Electromyography of biceps and quadriceps muscles was normal and did not reveal complex repetitive discharges. Evoked potentials revealed normal brainstem evoked potential and absent somatosensory evoked potentials from the tibial and median nerves. Visual evoked potentials were normal in the right eye (P100 – 101.40) and mildly prolonged in the left eye (P100 – 113.40)
In view of the skeletal markers, clinical suspicion of inherited neuropathy was initially considered. However, NGS showed a compound heterozygous mutation in the GAA gene. Subsequently, GAA levels in DBS showed a reduced total acid alpha-glucosidase level of 5.46 nmol/hr/ml (normal value - 10–60) and lysosomal acid alpha-glucosidase value of 1.22 nmol/hr/ml (normal value - 4.51–15.0), which confirmed the association. The pathological findings were as: on routine H & E stain, the muscle showed well preserved architecture with no significant variation in muscle fibre size or shape and with no distinct myopathic features. There were no vacuoles. The MGT stain and the enzyme histochemical stains failed to show any additional findings. EM also did not reveal storage deposits/inclusions and no organellar abnormality, including mitochondrial changes. The nerve findings indicated a significant loss of myelinated fibres (small and large) with a few regenerating axonal clusters/sprouts. EM revealed a very severe loss of myelinated fibres.
Case 5
A 35 year-old-lady presented with 10 years history of gradually progressive fatigable lower limb proximal muscle weakness. She had exertional dyspnea and orthopnea. She had mild respiratory difficulty at rest. Consequentially she also developed hypercapnia induced early morning headache. There was no diurnal fluctuation, myoglobinuria, myotonia, cramps, cranial nerve deficits, or sensory involvement. Examination revealed grade 4+ weakness of shoulder girdle and arms, grade 2–3 of the pelvic girdle and thigh. Tendon reflexes were normal. She had a waddling gait. She also has diabetes mellitus and hypothyroidism. Serum CK level was 1451 IU/L, SGOT of 146 U/L, SGPT of 102 U/L. Nerve conduction studies were normal with no decrement. Arterial blood gas showed increased PCO2 levels (80 mmHg). A pulmonary function test revealed a severe restrictive abnormality. 2D ECHO was normal. Total acid alpha glucosidase (12.87 nmol/hr/mL) was normal and lysosomal acid alpha-glucosidase (2.59 nmol/hr/mL) was significantly reduced.
Genetic results
NGS with clinical exome panel detected putative disease-causing GAA mutations in all 5 patients: homozygous variants in the first two cases and compound heterozygous in the remaining 3 cases (Table 1). While case 1 had a novel missense variant c.1461C > A (p.Phe487Leu) in a mutational hotspot region of exon 10, case 2 was found to have a reported variant c.1082C > T (p.Pro361Leu) in exon 7. Case 3 was compound heterozygous with a novel in-frame deletion in exon 14, c.1935_1940del (p.Val646_Cys647del) and a commonly reported intronic splice affecting variant c.-32-13T >G (intron 1). For case 4, one reported c.971C > T (p.Pro324Leu) and another novel c.794G > A (p.Ser265Asn) missense changes were identified in hot spot regions of exon 6 and 4, respectively. A reported insertion leading to frameshift c.1396dupG (p.Val466GlyfsTer40) and a reported synonymous variant c.546G > T (p.Thr182=) predicted to effect the splicing at 3’ end of exon 2 were detected in case 5. No other significant variants were identified in known neuromuscular disease genes in all the fivepatients.
Summary of the clinical phenotype, biochemical and mutation findings
#Not reported in Pompe mutational database. *Based of variant severity classification in Pompe mutational database (http://www.pompevariantdatabase.nl/).
DISCUSSION
Pompe disease was first described in 1932, and in 1959 Sant’Agnese included the presence of “marked cardiac enlargement and death in infancy” in the diagnostic criteria. Later, milder forms of acid maltase deficiency were discovered, and the first LOPD case was reported in 1968 by Engel and Dale [12]. Subsequently, short series and single case studies of LOPD have documented varied phenotypic presentations. LOPD usually presents as prominent proximal and axial muscle weakness, while distal involvement occurs in late stages. The involvement of respiratory muscles is early and profound in about a third of the cases [13]. In the current report, we present five genetically and enzymatically confirmed LOPD cases with varied and unusual phenotypes. One had LGMW with tongue fasciculations, minipolymyoclonus, and hyperactive tendon reflexes and respiratory distress, the second had rigid spine syndrome with gastrointestinal manifestations, the third had protracted LGMW with respiratory difficulty, ptosis, ophthalmoparesis, and episodic fever/exertion triggered profound muscle weakness, the fourth had progressive large fibre neuropathy, and the fifth had progressive limb-girdle syndrome with early respiratory involvement.
Mutational analysis identified eight unique potentially disease-causing GAA variants in our five patients. While five of these have been previously reported and functionally classified in Pompe variant database (http://www.pompevariantdatabase.nl/), three novel variants have been identified in cases 1, 3 and 4 respectively (Table 1) [14]. The mutational pattern of our patients is consistent with late-onset, less severe phenotypes, and all had a reduction in GAA activity. The intronic variant (c-32-13T > G) present in case 3 is a commonly reported LOPD variant, most often in the compound heterozygous state with variable phenotypes due to partial or complete skipping of exon 2 of GAAgene [8]. While case 5 had a frameshift variant in one allele, the synonymous variant c.546G > T (p.Thr182=) in the other allele is reported to cause a leaky splice site with intact transcript and residual GAA activity [15]. None of our patients had biallelic truncating mutations, which results in a complete loss of GAA activity. Among the reported variants except for one frameshift variant c.1396dupG (p.Val466GlyfsTer40) in case 5, all others have been predicted as having less severe to mild effect on GAA expression [14]. While case 1 had a novel homozygous missense variant c.1461C > A (p.Phe487Leu) in the hotspot region of exon 10, another missense change c.1460T > C (p.Phe487Ser) affecting the same codon has been previously reported as a less severe variant associated with Pompe disease [14]. Except for case 4, all other patients had at least one variant affecting the functional Glycosyl hydrolase domain between amino acid positions 340-824 of GAA (http://pfam.xfam.org/protein/P10253).
In a study from mainland China, among 27 patients of LOPD diagnosed by histopathology, the median age of onset was 21 years, and the majority had presented with proximal muscle weakness [16]. Our patients also had similar age at onset (except cases 2 and 4), and all had predominant proximal and axial muscle weakness. In another large cohort of 30 LOPD patients of Caucasian origin, the median age at onset and diagnosis was 31 and 40 years, respectively and with a diagnostic delay of 8.6 years. The majority had limb-girdle and axial muscle weakness. None had presentation as chronic anterior horn cell disease as recognized in our case 1 [17]. In comparison, our cases had a much earlier age of onset with unusual presentations along with the classical proximal muscle weakness. One case had progressive anterior horn cell involvement, one had rigid spine syndrome, third case had ocular and early respiratory involvement with LGMW, fourth case had peripheral neuropathy without muscle involvement.
In the infantile forms of Pompe disease, the muscle shows excessive vacuolations involving both fiber types, autophagy, and mitochondrial derangements. In the juvenile and adult-onset LOPD, the percentage of vacuolated fibers reduces to 50% and 10–15%, respectively [18] and are predominantly located in type 2 fibers. Rarely, LOPD can also demonstrate severe vacuolar myopathy involving type1 and 2 fibers, as observed in our first case.
The glycogen accumulation in spinal motor neurons leading to a swollen appearance in CNS has been documented in earlier studies [19, 20]. In a recent study in the mice models (Gaa–/–) of Pompe disease, authors have demonstrated glycogen accumulation in the phrenic motor neurons and cervical spinal cord. The electrophysiology and behavioral aspects of breathing in Gaa–/– mouse and transgenic line (GAA only in the muscles) proved that the insufficient neural output might be a possible cause for early respiratory involvement rather than muscle weakness. They found high glycogen content in the spinal cord (0.04%) compared to brain tissue (0.01%) in a patient who died due to progressive ventilatory insufficiency inspite of treatment with rhGAA from 6 months of age [21]. This might be the possible reason for considerable variability in the success of enzyme replacement therapy [22]. Glycogen accumulation with ongoing apoptosis was present in the medulla and spinal motor neurons even in a 6 week old asymptomatic Pompe (Gaa–/–) mice [23]. These studies implicate inevitable neurodegeneration and altered neuronal excitability due to glycogen aggregation in the motor neurons, even in the early stages of the disease process. Denervation potentials in EMG mimicking spinomuscular atrophy are reported in a single case [24]. Our first patient had features of progressive AHCD which could be supported by the above studies, but in addition, he had hyperactive tendon reflexes. AHC involvement has also been postulated by a monforte et al who described a patient of LOPD with extensive fasciculations.[25] A few reports have described the occurrence of dilative arteriopathy, basilar artery dolichoectasia and intraparenchymal hemorrhages in LOPD [26–28]. MRI of the brain in our first patient was normal.
Rigid spine syndrome (RSS) is a characteristic feature in primary muscle diseases like Emery-Dreifuss muscular dystrophy (EDMD) with LMNA mutations, SEPN1 related congenital muscular dystrophy and myopathy, FHL1 related myopathy and myofibrillar myopathy with BAG3 mutations. Pompe disease is not a classical differential diagnosis in RSS. Our patient 2 presented with the unusual phenotype of RSS which earlier has been described in very few case reports on LOPD. In these reports, LOPD presented as RSS with respiratory insufficiency, which was a prominent feature leading to the diagnosis. To the best of our knowledge, there are only two cases presenting with the progressive rigid spine as an initial symptom without respiratory insufficiency [29, 30].
Case 3 was also found to have subclinical hypothyroidism following treatment for thyrotoxicosis and its exacerbations. According to a study by Schneider et al.,hypothyroidism was found in 50% of Pompe disease patients compared with the University of Minnesota Medical Center adult patient population. Fluctuations in muscle strength have been associated with metabolic myopathies, but no such case has been reported with Pompe disease. Our patient 3 showed a peculiar feature of documented acute deterioration in his symptoms triggered by febrile illness / unusual physical activity followed by prolonged recovery over several weeks.
In a study from France, whole-body muscle MRI was performed in 20 patients of LOPD, and no consistent pattern of involvement was identified in thigh muscles, except for preserved rectus femoris, gracilis, and sartorius even in severe cases. Further, the tongue muscles, subscapularis, and lumbar extensors were prominently affected [31]. In another longitudinal study of 23 patients, MRI had revealed rapidly progressive fatty degeneration in thigh muscles compared to lower leg muscles with preferential involvement of adductor longus and magnus. Myoedema was found mainly in biceps femoris, semi-membranosus and adductor magnus [32]. Muscle MRI in patient 1 was dissimilar to the above reports at the first evaluation,but follow-up MRI demonstrated severe atrophy of thigh muscles and preferential involvement of posterior leg muscles which is similar to the above study. The second patient had exactly same findings as reported in the previous study. Patient 3 also showed similar findings with rectus femoris sparing.
Previously viewed as primary muscle disease, Pompe disease is now established as a multisystem disorder. Peripheral nerve involvement as a presentation of Pompe disease has been documented very infrequently. Autopsy reports have described the presence of glycogen deposits in Schwann cells [33, 34]. Two Pompe disease patients with clinical evidence of small fiber nerve involvement are reported among a small case series. In the same report, the authors also screened 44 consecutive Pompe disease patients for small fiber nerve involvement and found that 50% of patients were at risk [35]. Monteiro et al., recently reported 4 cases of Pompe disease with concomitant large fiber involvement suggesting potential peripheral nerve involvement in Pompe disease [36].
Our case 4 did not show any symptoms suggestive of muscle involvement and rather presented with a childhood-onset sensory-motor polyneuropathy associated with skeletal markers mimicking inherited neuropathy. LOPD was suspected retrospectively in him following NGS result showing putative disease-causing compound heterozygous variants in GAA gene. Subsequent measurement of GAA activity confirmed the diagnosis of LOPD. This is probably the first case of LOPD presenting as polyneuropathy without clinical features suggestive of primary muscle involvement.
As we have described above Pompe disease can mimic various neuromuscular disorders, one of them being LGMDs. Prevalence of LOPD in unclassified LGMD varies from 0.8–4.6% across various studies [37–40]. Preisler et al., determined a prevalence of LOPD amongst unclassified LGMD patients of as high as 8% [41]. Evidence suggests that LOPD shall be considered as a differential in patients presenting with LGMD pattern of weakness, especially when they show red flags such as axial and respiratory involvement that is out of proportion to that of limb involvement, mild non-dystrophic myopathic features on muscle biopsy, and CK levels < 1000 Units/L. Case 3 and 5 both presented with a prolonged history of LGMD pattern of weakness with evidence of prominent respiratory muscle involvement, which gave a clue towards the diagnosis.
ERT with alglucosidasealfa was approved in 2006 [42] for all patients of pompes disease based on robust data suggesting benefit in IOPD, though only small uncontrolled studies demonstrated benefit in LOPD. The first randomized controlled trial LOTS (Late-onset Treatment study) in LOPD demonstrated an improvement in walking distance and stabilization of respiratory function [43]. Efficacy of ERT in LOPD is difficult to evaluate due to the clinical heterogeneity and small sample size of the published studies. Therefore various systematic reviews have been conducted. [44–46] and the recent meta-analysis conducted by Sarah et al. [47] revealed that the ERT leads to an improvement in the walking distance but there is no statistically significant improvement noticed in the muscle strength or respiratory function, although a trend towards an increase in muscle strength was seen, five fold lower mortality rate was reported in a systematic review by Schoser et al. [45] Based on these results, American association of Neuromuscular and Electrodiagnostic Medicine [47] recommends ERT in all confirmed and symptomatic patients and the European Pompe Consortium [48] also recommends ERT in symptomatic patients who give consent for compliance and regular monitoring and who have residual skeletal and respiratory muscle function, and without any advanced life threatening illness.
In a recent article, Angelini has shown that among 58 of 65 patients with LOPD, the only clinical parameters that were significantly associated with the responder category were pre-ERT walking distance and the use of regular diet, exercise, or both. Hence along with ERT, concomitant diet and aerobic exercise therapy are beneficial [49].
Patient 1 for the last 3 years has been provided with ERT through a local governmental support. ERT was advised to all other patients as well but as they belong to different states in India and due to the high cost it could not be initiated. All patients were advised for appropriate physical/ occupational therapy along with vitamin D and calcium supplementation.
In conclusion, it is important to recognize rarer non-muscle phenotypes of LOPD, which would define its vast spectrum, natural history, and prognosis. The varied spectrum of LOPD presentations as observed in our patients often poses difficulty in diagnosis and management. However, with availability of NGS and ease of screening large number of genes using targeted panel or exome sequencing techniques, prompt genetic testing and reverse phenotype correlation can be valuable in early identification of unusual LOPD cases. Although an early diagnosis of LOPD permits prompt intervention with ERT, the probable manifestations of CNS and its response to therapy are not clear.
Footnotes
ACKNOWLEDGMENTS
We would like to thank the patients and their families for participating in this study.
CONFLICTS OF INTEREST
No potential conflicts of interest to declare.
FUNDING
The authors did not receive any financial support for this article’s research, authorship, and/or publication.