
Abstract
Hereditary transthyretin amyloidosis (hATTR amyloidosis) is a multisystemic disease usually presenting in a mixed neurological and cardiological phenotype. We present a case of hATTR amyloidosis associated with Leu55Arg mutation causing a form of familial oculo-leptomeningeal amyloidosis. Two brothers and their mother presented with severe autonomic neuropathy, loss of visual acuity and lepto-meningeal involvement. One patient suffered subarachnoid hemorrhage as a possible complication of cerebral involvement. The patients suffered from treatment-refractory weight loss and recurring vitreous opacities. RNA interference-based treatment has led to stabilization of autonomic and peripheral neuropathy but has had no effect on ocular symptoms.
Introduction
Hereditary transthyretin amyloidosis (hATTR amyloidosis) can cause a wide range of symptoms, most importantly an axonal sensorimotor polyneuropathy (familial amyloid polyneuropathy, FAP) and cardiomyopathy. Ocular, renal and cerebral involvement is also well established in the context of this disease [1, 2]. We present the cases of two brothers affected by oculo-leptomeningeal transthyretin amyloidosis associated with a Leu55Arg mutation in the TTR gene and a positive family history for amyloidosis. Up to date, this rare mutation has only been reported in mainland Chinese families affected by vitreous amyloidosis and polyneuropathy [3, 4].
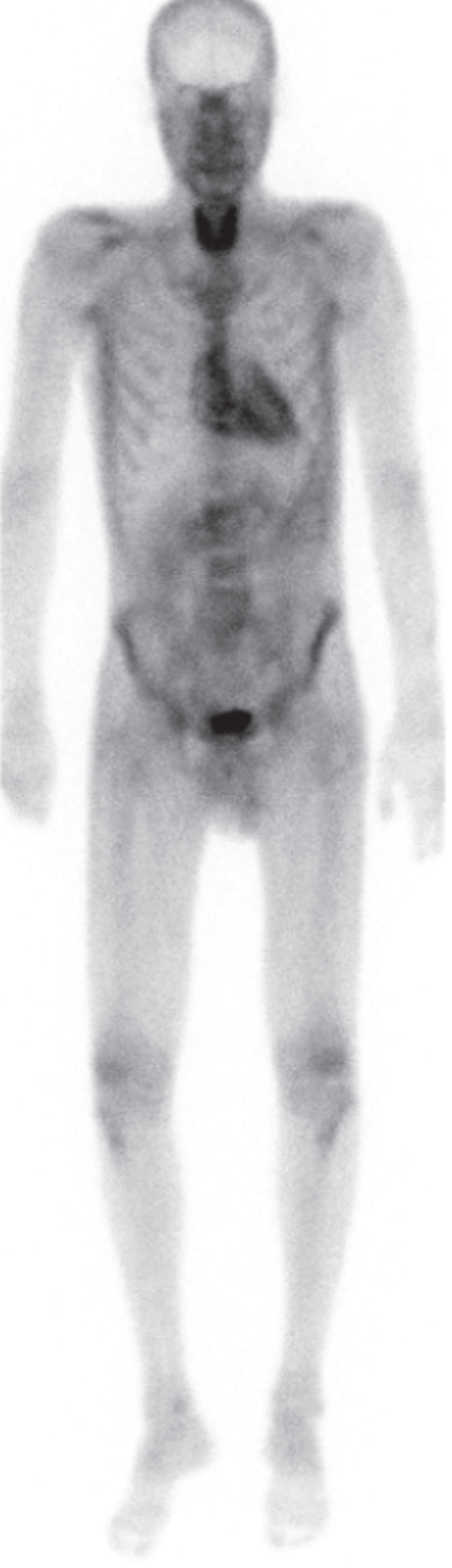
DPD scintigraphy of patient A: Whole body planar image 3 hours post-injection showing mild cardiac and extracardiac enhancement. Note the enhancement in the thyroid gland.
Patient A
The 35-year-old patient initially presented in 2016 with painful paresthesia in his arms and legs, vertigo, unintentional weight loss, diarrhea and erectile dysfunction. The patient also reported a loss of visual acuity. The symptoms had started approximately two years earlier. The initial diagnostic workup included skin biopsy, colonoscopy/gastroscopy and neurological, cardiological and ophtalmological evaluation. The skin biopsy showed a reduced intraepidermal nerve fiber density (IEND) consistent with small fiber neuropathy (SFN). The congo-red staining was negative. The gastric biopsies, however, were positive for congo-red material with green birefringence under polarized light. The neurological examination showed FAP stage 1 neuropathy with distal-symmetric, painful sensory neuropathy with sensory ataxia. Electrophysiological exams demonstrated a mild sensory-motor, axonal demyelinating polyneuropathy in the lower limbs. The upper limbs were not affected and there was no sign of carpal tunnel syndrome. Cardiological examinations showed a normal ECG; echocardiography revealed mild left ventricular (LV) hypertrophy with normal apical strain. Genetic sequencing of the TTR gene showed heterozygous Leu55Arg-Mutation (c224.T>G). Two months later, the patient presented to the emergency department with aphasia and severe headache. Diagnostic workup showed a CT-negative subarachnoid hemorrhage (SAH) with no apparent source of bleeding (e.g. aneurysm). CSF analysis showed xantochromia, pleocytosis and elevated protein. The patient recovered quickly from this episode. We started treatment with Tafamidis one month after the initial presentation. Due to progressive worsening of visual acuity, the patient subsequently underwent bilateral pars plana vitrectomy. We switched treatment to Patisiran two years after initial presentation due to the progression of the severe autonomic neuropathy (progressive orthostasis, dramatic weight loss). Follow up diagnostic workup showed a grade 2 99mTc-Dicarboxi-Propan-Diphosphonat (DPD) Scintigraphy with unusual enhancement in the thyroid, stable LV hypertrophy and normal ECG (see image 1). The MRI showed a circumferential cervico-thoracal leptomeningeal enhancement and mild hydrocephalus, the latter as sequelae of the SAH (see image 3). Neuropsychological testing detected mild cognitive impairment in the domains attention, working/short-term memory and information processing speed. The patient has received Cannabinoid-based treatment for neuropathic pain for 2 years. Until today, he has well tolerated the treatment with Patisiran. Polyneuropathy has stabilized; the patient has gained weight and reported less autonomic symptoms. Weight gain was observed 12 months after initiation of treatment with Patisiran. Ophthalmological follow up examinations have indicated further progress of vitreous opacities and loss of visual acuity.
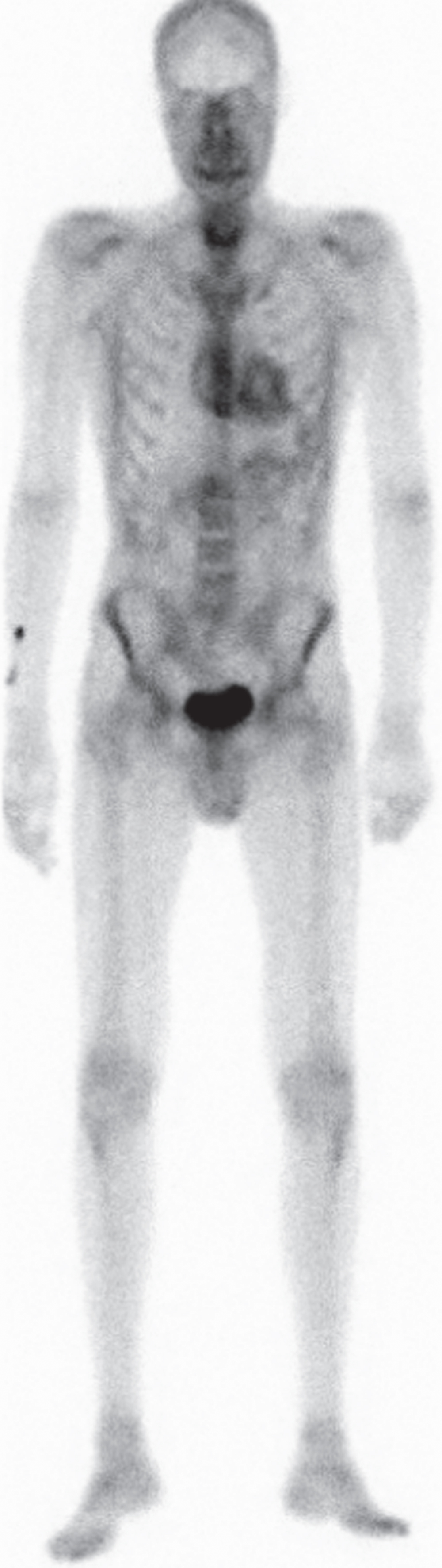
DPD scintigraphy of patient B: Whole body planar image 3 hours post-injection showing the same enhancement pattern as seen in Fig. 1.
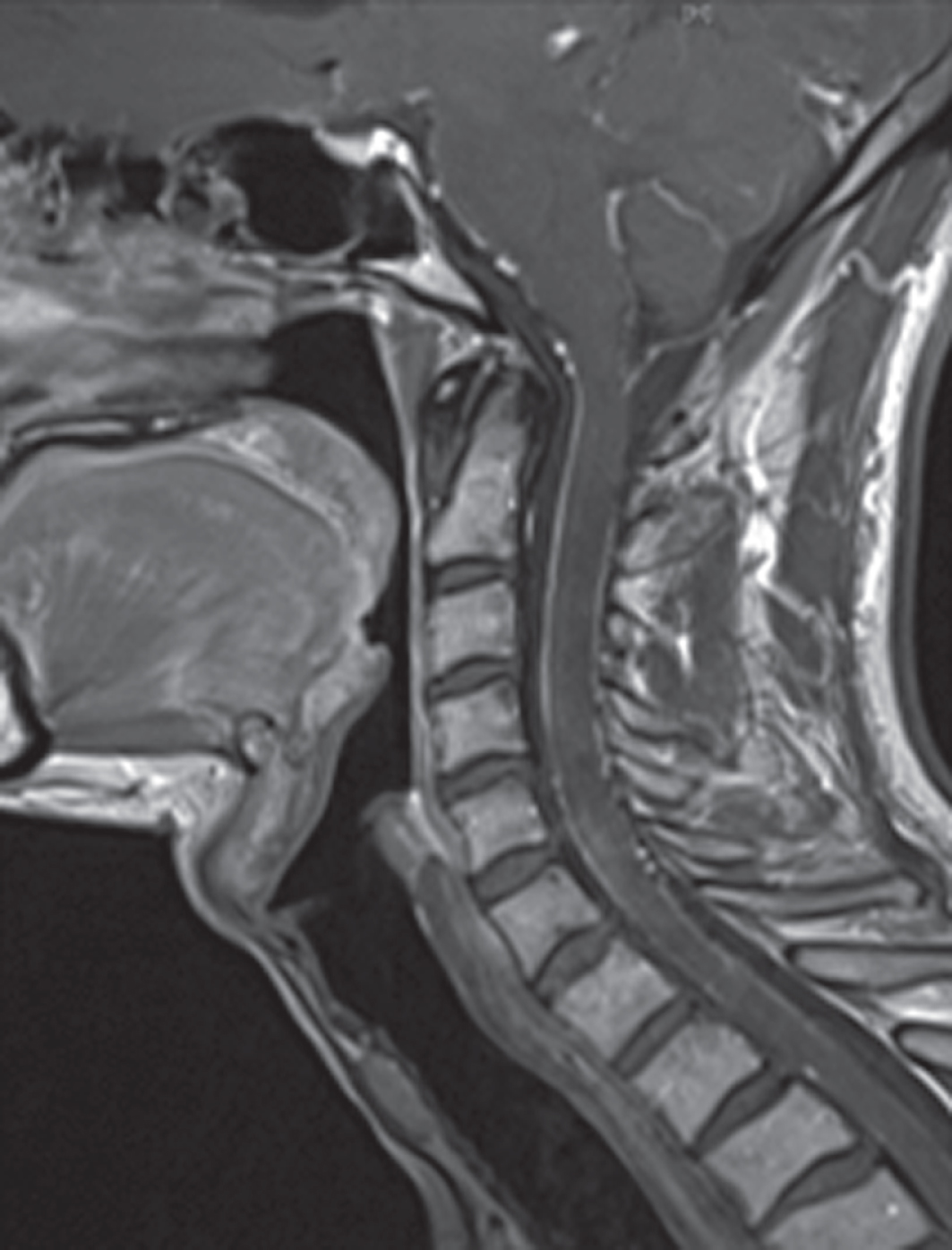
Contrast-enhanced MRI of patient A: circumferential cervico-thoracal leptomeningeal enhancement.
Patient B
The 39-year-old brother of patient A initially presented to an ophthalmologist, complaining about a loss of visual acuity. He was diagnosed with vitreous opacity of unknown etiology. The patient subsequently presented to an urologist because of erectile dysfunction. At that time, he also reported fatigue and an unintentional weight loss of around 20 kilograms in the two previous years. He was referred to a gastroenterologist and underwent colonoscopy and gastroscopy, where multiple biopsies were obtained. Histology showed congophilic material and immunohistochemistry was positive for transthyretin. The patient was referred to the department of neurology in 2016. The initial neurological examination showed painful distal-symmetric sensory neuropathy with sensory ataxia. After an electrophysiological examination, we diagnosed a mild sensory-motor, axonal demyelinating polyneuropathy in the lower limbs, FAP stage 1. Echocardiography showed only mild LV hypertrophy without apical sparing, the ECG was normal. The MRI showed no signs of leptomeningeal enhancement.
Genetic sequencing of the TTR gene detected the same heterozygous Leu55Arg-Mutation (c224.T>G) in the TTR gene as in case A. We initiated treatment with Tafamidis. The patient subsequently underwent bilateral pars plana vitrectomy due to progressive vitreous opacities in both eyes. We saw a further disease progression with dramatic weight loss and progression of orthostasis. We therefore started treatment with Patisiran one and a half years after initiation of Tafamidis treatment. We performed a DPD Scan at baseline of Patisiran treatment that showed Perugini grade 2 and, as in case A, notable enhancement in the thyroid gland (see image 2). The patient has well tolerated the treatment with Patisiran. Peripheral neuropathy has stabilized whereas autonomic symptoms have slightly improved: 15 months after initiation of treatment, the patient has started to gain weight. Unfortunately, recent ophthalmological follow up examinations showed a progression of vitreous opacities.
Discussion
We presented two cases of early-onset hATTR amyloidosis associated with Leu55Arg mutation. At the time of presentation of case B, the mother of the two brothers reported in case A and B had already died at age 56. She was also a symptomatic carrier of the Leu55Arg mutation and had undergone a liver transplantation. She died of postoperative heart failure caused by amyloid cardiomyopathy. She suffered from distal-symmetric and autonomic neuropathy, bilateral carpal tunnel syndrome and visual impairment. The similarities in clinical presentation of case A and B are remarkable: both patients presented in their mid-30s with loss of visual acuity and dramatic weight loss caused by severe autonomic neuropathy. In contrast to that, the severity of peripheral neuropathy is only mild in both patients. Nevertheless, we saw relevant and painful Small Fiber Neuropathy in both cases. Cardiac involvement is present in both echocardiography and DPD scans, although up to today, there have been no clinical findings suggestive of heart failure. Nevertheless, the mother of the two patients died of cardiac complications at age 56. Patient A suffered a SAH of unknown origin. As cerebral (leptomeningeal) involvement was obvious in the MRI of this patient, SAH might have been caused by cerebral amyloid angiopathy. Another possible explanation is the use of Sildenafil, used for the treatment of ED in this patient. Interestingly, DPD scans of both patients showed an unusual enhancement in the thyroid gland in the absence of thyroid dysfunction. The significance of this finding remains unknown. Long et al. published an ophthalmological case series of Chinese hATTR amyloidosis patients of one family with Leu55Arg mutation [3]. The authors described an early-onset phenotype characterized by severe ocular amyloidosis and, in some of their patients, chronic diarrhea. They did not report the neurological and cardiac phenotype of their patients. Another Chinese case series reported ocular amyloidosis in Gly67Glu-associated hATTR amyloidosis, again without specifying the neurological phenotype [5]. Fan et al. reported a case series of a Chinese family with exclusive leptomeningeal amyloidosis without vitreous opacities and neuropathy [6]. Therefore, our case report is the first to comprehensively describe the phenotype of Leu55Arg-associated hATTR amyloidosis outside the Chinese mainland.
In summary, Leu55Arg-associated hATTR amyloidosis can present with a mixed, predominantly oculo-leptomeningeal and autonomic phenotype. Painful Small Fiber Neuropathy has a relevant impact on the quality of life in these patients. Intracerebral hemorrhage/SAH may be a rare complication of oculo-leptomeningeal amyloidosis. Although clinically significant cardiomyopathy might initially not be present in Leu55Arg-associated hATTR amyloidosis, patients might develop severe cardiomyopathy later in the course of the disease. Treatment with novel therapeutic agents (Patisiran) seems to stabilize peripheral neuropathy and autonomic symptoms (BMI) but has had no effect on ocular symptoms in our patients. Nutritional status might indeed be an early indicator of treatment response to Patisiran, as already suggested by phase-3 trial and first real world data [7, 8].
Conflicts of interest
There are no conflicts of interest to declare.