
Abstract
The high incidence of mutations and the crucial roles of KAT2A in cancer development have received increased attention. Nevertheless, a systematic comparison of the heterogeneity and dynamics across different cancer types has not been conducted. Hence, a deep analysis using public databases was performed to clarify the contributions of KAT2A and its correlation with tumorigenesis. The raw data regarding KAT2A expression in cancer patients and healthy controls were obtained from The Cancer Genome Atlas (TCGA). Sexually dimorphic manner, genomic alterations, and expression pattern of KAT2A, as well as the association of the KAT2A with survival, were retrieved from UALCAN, cBioportal, and TISIDB databases. Additionally, the Protein-Protein Interaction (PPI) analysis was conducted using the STRING database. The human protein atlas was used to obtain the staining results of protein levels in cancer and normal samples. The correlation between KAT2A and its potential target drugs was determined using TISIDB and HISTome2. Compared to the normal tissues, CHOL and TGCT tumors presented significantly high KAT2A expression, which was positively correlated with BLCA, BRCA, CESC, CHOL, COAD, ESCA, HNSC, KICH, KIRP, LIHC, LUAD, LUSC, READ, STAD, and THCA. However, no significant difference was detected between normal and tumor tissues for the sex difference pattern of KAT2A expression. The PPI analysis indicated that TADA3, CCDC101, TRRAP, SUPT3H, MYC, TADA2A, and USP22 levels were positively correlated with KAT2A expression, while TADA2B and ATXN7 were negatively correlated. A positive link of KAT2A with cancer isotypes and significant connections of the KAT2A expression to poor overall and disease-free survival were also observed. Further validation was conducted using immunohistochemistry (IHC) staining, qPCR, and Western blot. Some potential HAT inhibitory drugs of KAT2A were also determined, but more work and clinical trials are required before their application.
Keywords
Introduction
Historically, cancer remains the leading cause of death, posing a significant threat to human health and lifespan on a global scale. According to World Cancer Research Fund International data, there were an estimated 18.1 million cancer cases worldwide in 2020; breast and lung cancers were the most common, contributing 12.5 and 12.2% of the total number of new cases diagnosed [1]. Generally, the young population has a relatively lower cancer incidence than older people, and cancer incidence rises dramatically with age [2].
Lysine acetyltransferase 2A (KAT2A), or General control nonderepressible-5 (GCN5), is a histone acetyltransferase (HAT) that primarily functions as a transcriptional activator. Its complex belongs to the GNAT (Gcn5-related N-acetyltransferase) family of acetyltransferases and are transcription co-activators [3]. KAT2A also functions as a repressor of NF-
Histone acetylation by HAT gives a specific tag for epigenetic transcription activation [8] and further destabilizes nucleosomes by promoting the dissociation of H2A-H2B dimers from nucleosomes for nucleosome assembly, DNA damage repair, and transcriptional regulation [12]. Accordingly, KAT2A also participates in the regulation of post-translational modifications by the acetylation of nonhistone proteins, such as CEBPB (CCAAT/enhancer binding protein beta), PLK4, and TBX5 (T-box transcription factor) [13]. Emerging evidence suggests that KAT2A is differentially expressed in metabolic disorders and is crucial in multiple human diseases, including cancers [14, 15]. KAT2A protein levels are significantly upregulated in urothelial cancer cell lines and human colon cancer tissues compared to normal controls [16, 17]. Recently, a research group in MD Anderson Cancer Center has identified KAT2A as a critical coactivator of cell-cycle gene expression driven by MYC overexpression, with significant implications for B-cell lymphomagenesis. The depletion of the GCN5 gene is notably associated with MYC-derived B-cell lymphoma, highlighting the potential therapeutic value of targeting KAT2A as a viable drug option in cancer treatment [18]. Hu et al. suggested that GCN5 and PCAF mediated acetylation of RAE-1 protein can activate NKG2D-dependent immune surveillance within HEK 293 cells and GCN5 and PCAF knockdown in osteosarcoma and lung cancer result in impaired induction of the natural killer group 2D (NKG2D) ligand Rae-1 [19, 20]. In head and neck squamous cell carcinoma, H3K27 acetylation, a GCN5 target, activates PD-L1 and galectin-9 transcription to evade tumor immunity [21].
Some studies have explored the role of KAT2A in malignancies. However, the effect of KAT2A expression and its association with tumorigenesis remains unknown. Cancer immunotherapy, especially immune checkpoint blocking (ICB), has emerged as a prominent approach in cancer treatment. The identification of immunotherapy targets can be achieved via pan-cancer expression analysis. Therefore, exploring the correlation between target genes, clinical prognosis, and associated signaling pathways using public databases is of great interest. Given the intricacy of tumorigenesis, investigating the expression of potential target genes and the underlying molecular mechanisms in various cancers is crucial. Therefore, our main objective is to offer a comprehensive understanding of KAT2A gene expression and its correlations across different tumors. Additionally, we aim to explore the potential therapeutic value of KAT2A as a promising drug target for these cancers.
Material and methods
Human tissue source and ethical statement
This study was approved by the clinical trial ethical committee of the Shanghai Pulmonary Hospital, Tongji University School of Medicine. After the approval of the ethical committee, all methods and information collection in this study were performed according to relevant guidelines and regulations. This research complies with all relevant ethical principles. Samples were sent to Tongji University School of Medicine for further pathological analysis. All patient samples were collected following local regulations and after obtaining the informed consent approved by the institutional review board (IRB). This commitment to ethical guidelines ensures that the research respects the rights and well-being of the study participants. Details regarding the human tissues collected for this study are presented in Table 1.
Summary of human tissues used in the study
Summary of human tissues used in the study
RNA isolation (#74004, RNeasy Micro Kit, QIAGEN) and cDNA preparation (#QP057, SureScript™ First-Strand cDNA Synthesis Kit, GeneCopoeia) were conducted following the manufacturer’s instructions. The qPCR using PowerUp™ SYBR™ Green Master Mix (#A25742, Thermo Fisher, Carlsbad, CA, USA) was carried out on the Bio-Rad CFX96 system (#3600037, Bio-Rad Laboratories). Results were normalized by the GAPDH gene and the mRNA levels of KAT2A were calculated versus GAPDH (2
Western blot
Protein extraction, quantification, and Western blot analysis were conducted following the manufacturer’s instructions. Briefly, tissue protein was mechanically dissociated using Dounce Tissue Grinders (D8938, Sigma-Aldrich, USA) and disrupters (TissueLyser LT, Cat. No.: 85600, QIGEN) at 4
Immunohistochemistry (IHC)
Samples were fixed in formalin and embedded in paraffin. The staining process was performed following the manufacturer’s instructions. Briefly, after antigen retrieval, the sections were treated with 3% H
Data source
UCSC Xena. The RNA sequencing (RNA-seq) expression profile and survival data regarding pan-cancer (33 cancer types) were downloaded from the University of California SANTA CRUZ database (UCSC Xena
Name and abbreviation of the cancers analyzed in this study
Name and abbreviation of the cancers analyzed in this study
Data source:
TISIDB database. TISIDB (
cBioPortal. The cBioPortal (
UALCAN. The UALCAN (
STRING. The STRING database (
Human protein atlas (HPA). The HPA (
HISTome2. The HISTome Infobase (
Data from individual experiments are presented as means
Sample numbers of all cancer types
Sample numbers of all cancer types
Transcriptional level analysis of KAT2A among human cancers
To determine the KAT2A gene expression in different cancers, we analyzed 11,014 samples (9,675 primary tumors, 719 solid tissue normal, 394 metastatic tumors, 173 primary blood-derived cancers, 42 recurrent tumors, 10 additional new primary tumors, and one additional metastatic tumor) spanning 32 different human tumors (Tables 2 and 3). We explored KAT2A mRNA levels using the UCSC Xena database (
Pan-cancer analysis of KAT2A gene expression. A. KAT2A expression in different cancer types from TCGA database. B. Radar map of KAT2A expression. C. KAT2A expression in various cancers compared to normal tissues. D–J. KAT2A expression decreases in BRCA, KIRC, LIHC, LUAC, LUSC, PRAD, and THCA. X-axis, sample types. Y-axis, transcript per million [log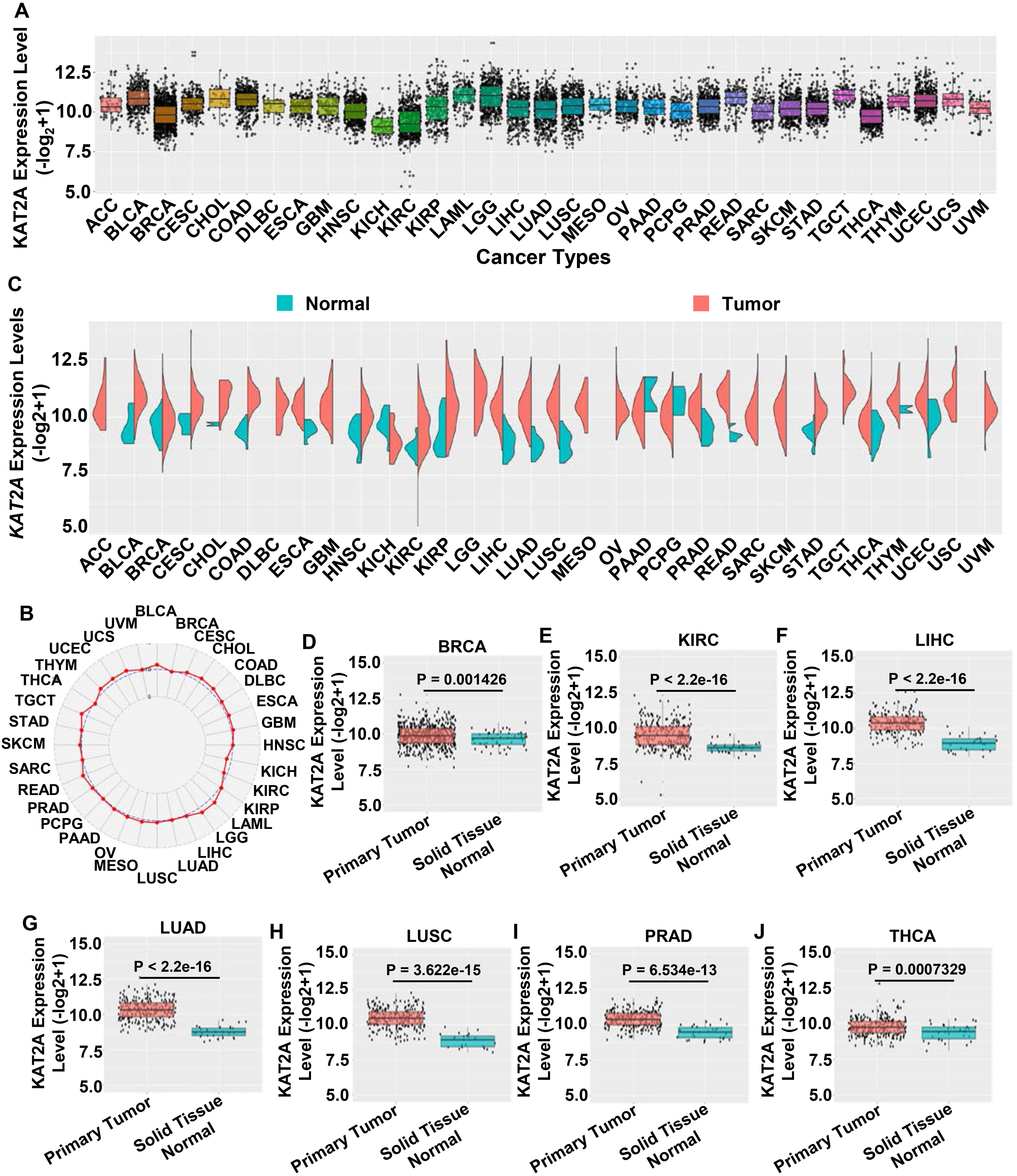
Cancer incidence has a sexually dimorphic pattern, making the underlying mechanisms a crucial issue in cancer research and the development of precision medicine [32]. This sexual dimorphism might result from biological sex differences (e.g., hormone discrepancies and/or chromosomal homo/heterogeneity) [33, 34], as well as lifestyle and habit, which are also connected to the incidence of cancers [35]. Therefore, precision medicine based on gender is an important modality [36, 37]. Furthermore, the National Institutes of Health (NIH) emphasizes studying male and female vertebrate animals and humans. In this study, we investigated whether KAT2A expression has a sex-specific pattern. First, we conducted a side-by-side analysis of the KAT2A mRNA levels in males and females using samples of additional new primary tumor, additional metastatic tumor, metastatic tumor, primary blood-derived cancer, primary tumor, recurrent tumor, and solid tissue normal. However, we did not detect significant differences between males and females (Fig. 2A). Then, we evaluated the sex difference expression pattern of KAT2A in normal (Fig. 2B) and tumor tissues (Fig. 2C) separately and did not detect significant differences. To gain further insights into KAT2A expression, we selected the top 5 cancers according to the
KAT2A gene expression gender bias across different cancers. A. Expression profile of the KAT2A gene between males and females based on sample types. B–C. Sex difference expression pattern of KAT2A in normal (B) and tumor tissues (C). D–H. KAT2A gene expression in READ (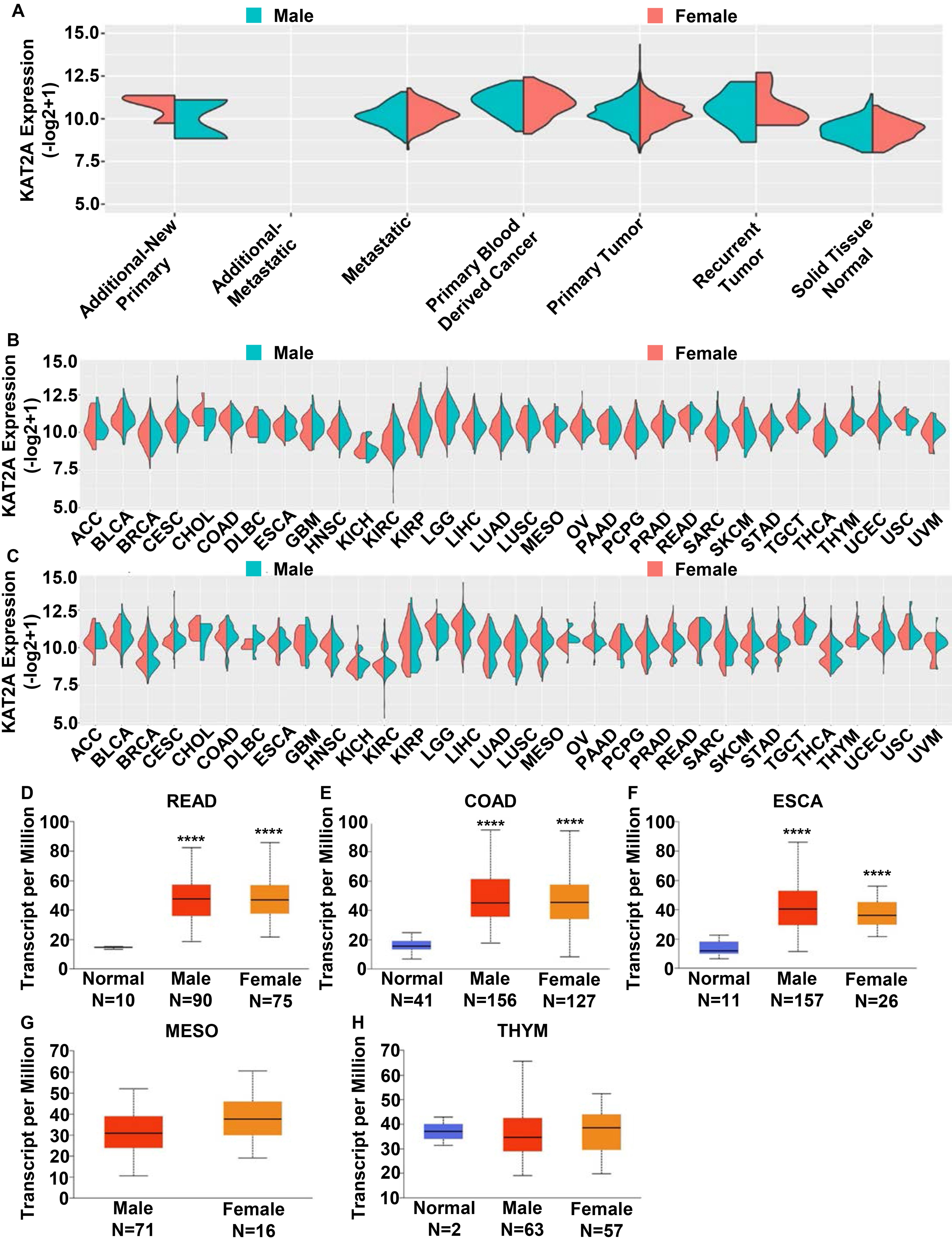
Expression of KAT2A gene based on individual pathological stages. A–O. KAT2A expression in different pathological stages (stages I-IV) of BLCA, BRCA, CESC, CHOL, COAD, ESCA, LIHC, KICH, KIRP, HNSC, LUAD, LUSC, READ, STAD, and THCA based on the UALCAN database. 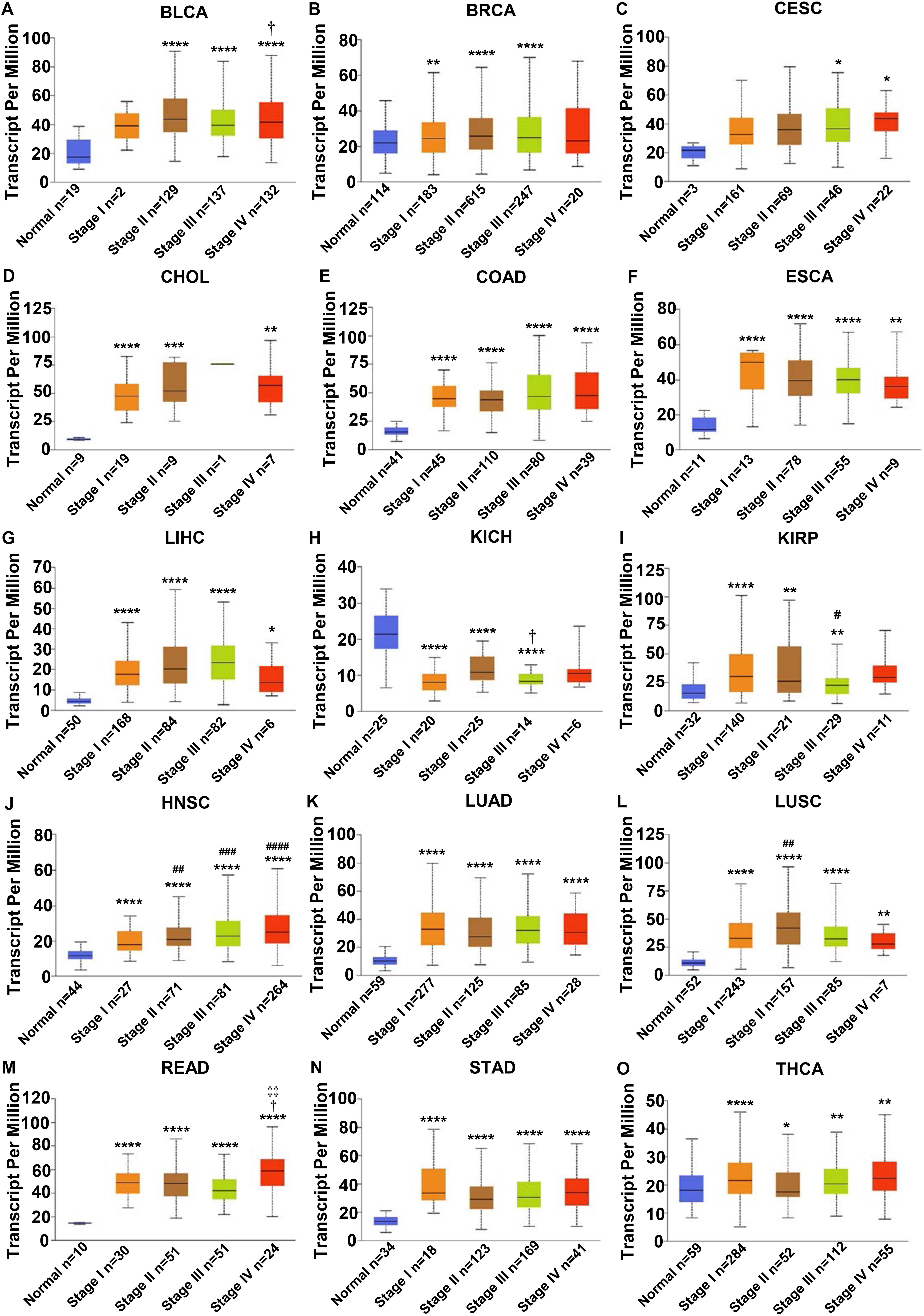
KAT2A gene interaction networks and correlation. A. Protein–protein interaction (PPI) network of the KAT2A-interacted proteins from the STRING website. B–K. Correlation between the expression of KAT2A and its related genes including USP22, CCDC101, MYC, SUPT3H, TADA2A, TADA3, TAF10, TRRAP, TADA2B, and ATXN7. 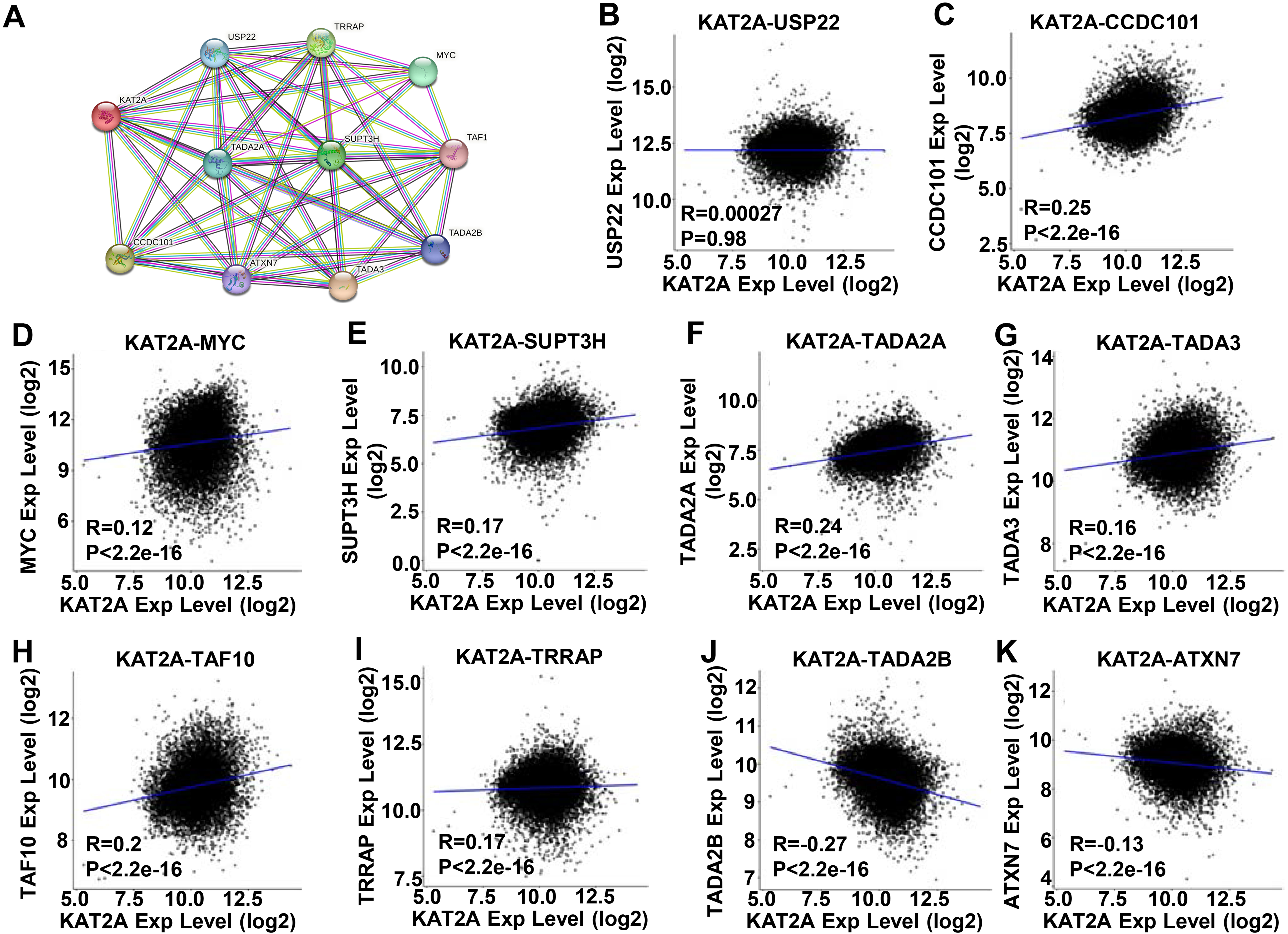
To assess the clinical relevance of KAT2A expression in pan-cancer, we investigated its correlation with cancer pathological stages. KAT2A expression was significantly correlated with pathological stages for BLCA (Fig. 3A), BRCA (Fig. 3B), CESC (Fig. 3C), CHOL (Fig. 3D), COAD (Fig. 3E), ESCA (Fig. 3F), HNSC (Fig. 3J), KICH (Fig. 3H), KIRP (Fig. 3I), LIHC (Fig. 3G), LUAD (Fig. 3K), LUSC (Fig. 3L), READ (Fig. 3M), STAD (Fig. 3N), and THCA (Fig. 3O) (
PPI correlation analysis of KAT2A
KAT2A is a subunit of the HAT module, which belongs to two distinct macromolecular complexes: the Human SAGA complex [38] and ATAC [39]. Thus, KAT2A is involved in a network that correlates with pathological tumor development. Thus, we evaluated the connections among the genes in this network using the STRING online database. The PPI network presented 10 nodes (TADA3, CCDC101, TRRAP, SUPT3H, MYC, TADA2A, USP22, TADA2B, ATXN7, and TAF10) involved in the functional interactions with KAT2A (Fig. 4A). Moreover, eight genes – USP22 (Fig. 4B), CCDC101 (Fig. 4C), MYC (Fig. 4D), SUPT3H (Fig. 4E), TADA2A (Fig. 4F), TADA3 (Fig. 4G), TRRAP (Fig. 4I), and TAF10 (Fig. 4H) were positively correlated with KAT2A. The remaining two genes, TADA2B (Fig. 4J) and ATXN7 (Fig. 4K) were negatively correlated with KAT2A. The function of these KAT2A-related proteins is summarized in Table 4.
Role of KAT2A-associated proteins in cancer development
Role of KAT2A-associated proteins in cancer development
Genomic mutation is closely associated with tumorigenesis [40, 41]. Thus, to identify mutation sites in KAT2A genes, we assessed 10,967 samples from 10,953 patients. As the lollipop plot depicted (Fig. 5A, post-translational modification (PTM) sites were frequently found in the p300/CBP-associated factor (PCAF) domain (86-337), Acetyltransferase domain (547-628), and bromodomain (738-818). The PCAF, acetyltransferase, and bromodomain in KAT2A are highly conserved across species [7, 42]. The types and sites of KAT2A genetic alterations are presented in Fig. 5A. We detected eight phosphorylation sites, six acetylation sites, one ubiquitination site, two methylation sites, and three SUMOylation sites for all 837 amino acids. The mutation sites contained 114 missenses, 16 truncating, five inframe, and four fusion mutations; R783Q was the most frequent (Fig. 5A).
KAT2A gene mutation across TCGA. A. Mutation site of KAT2A in different cancer types across protein domains. B. The alteration frequency of KAT2A in pan-cancer datasets according to the cBioPortal database. C. Mutation count of KAT2A across TCGA. 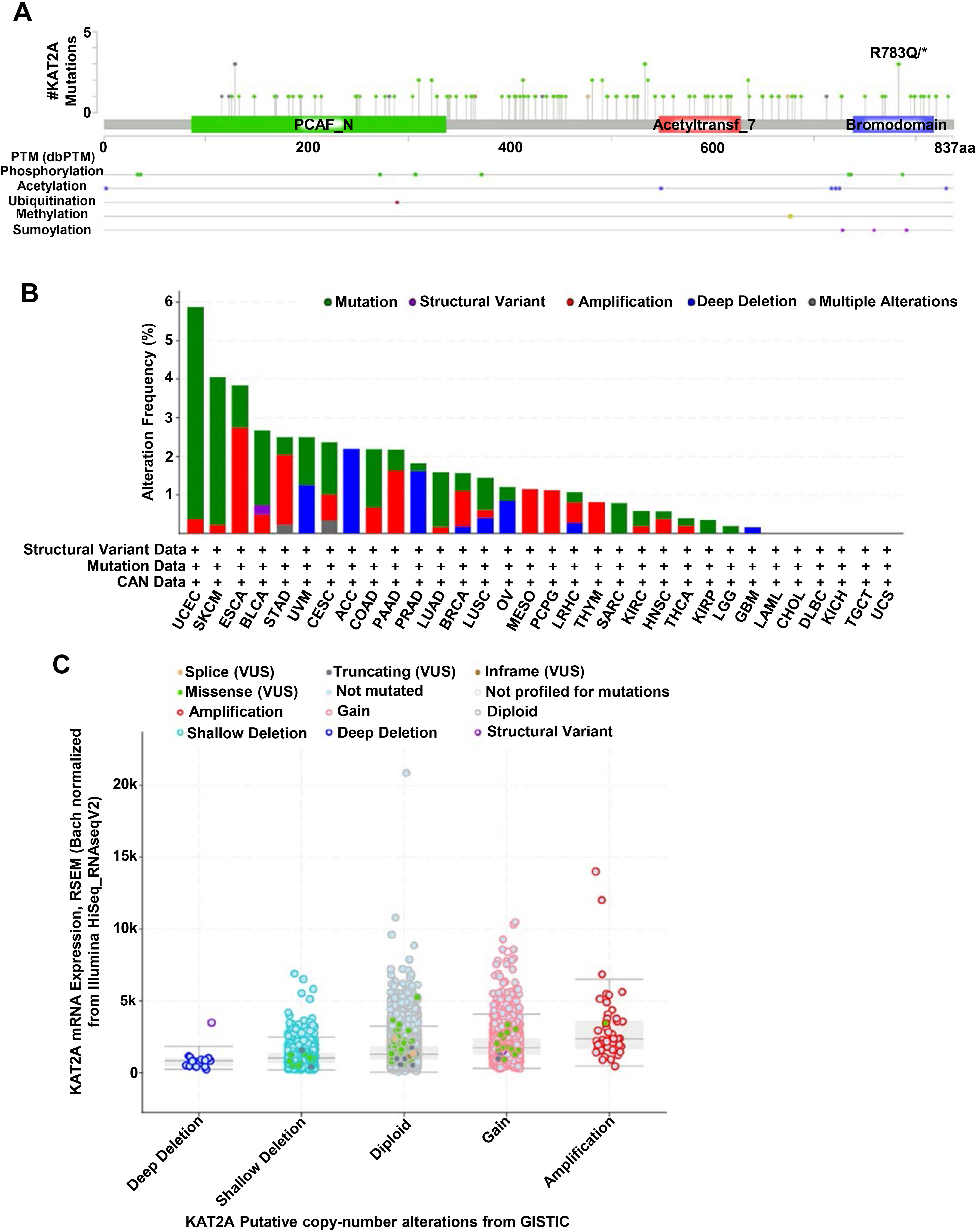
Next, we conducted a comparative analysis to determine the genetic alteration recurrence of the KAT2A gene in cancers. Then, we checked the genetic alterations of the KAT2A gene in cancer patients using the cBioPortal database. The frequency of KAT2A alteration significantly fluctuated across different cancers. The genetic alteration profiling of KAT2A showed that its mutation was one of the most important single factors for alteration in UCEC and SKCM. Also, we found the highest amplification frequencies of KAT2A in ESCA, STAD, PAAD, MESO, and PCPD (Fig. 5B).
Additionally, we analyzed the copy number alterations of the KAT2A gene among these cancers. We determined the highest amplification and the lowest deep deletion levels based on the batch normalized from Illumina HiSeq analysis, indicating a relatively rare frequency (Fig. 5C). Hence, various genetic alterations were implicated in KAT2A and might contribute to cancer development.
As a significant part of the complex microenvironment, the KAT2A-contained SAGA complex is significantly connected to the development and progression of various cancers [43, 44]. The quantity and activity status of tumor-infiltrating lymphocytes are important predictive criteria for cancer survival times [45, 46]. Cytotoxic CD8
Immune correlation analysis of KAT2A across TCGA. A. Correlation of KAT2A expression with immune infiltrating cells in the TISIDB database. B–H. Correlation of KAT2A expression in BRCA, KIRC, LIHC, LUAD, LUSC, PRAD, and THCA with infiltrating levels of act_CD8+ T cell. 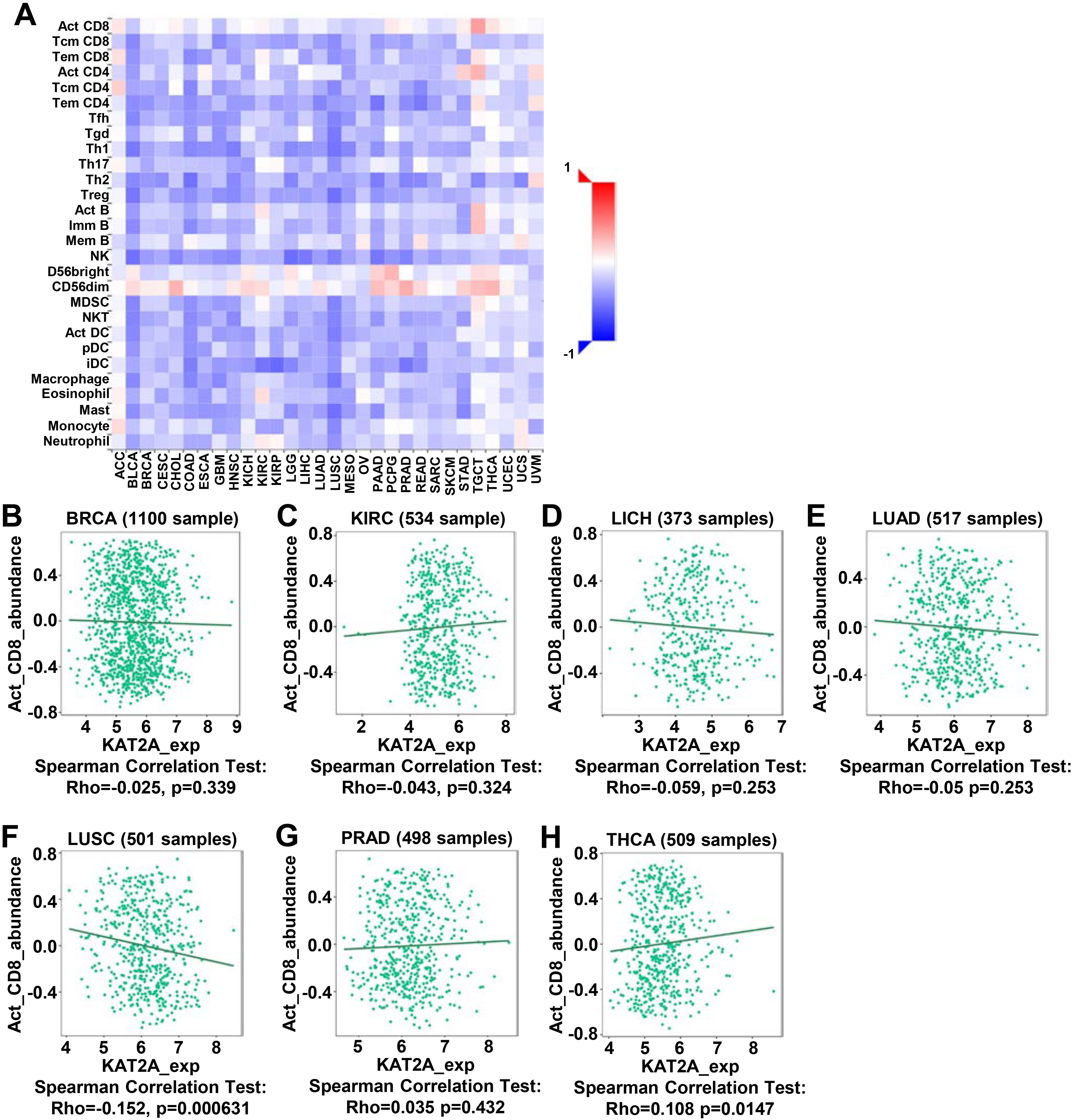
Our analysis using TCGA data showed that compared to the normal control tissues, BRCA, KIRC, LIHC, LUAC, LUSC, PRAD, and THCA tissues have significantly elevated KAT2A expression and that KAT2A expression is correlated with the infiltration of CD8 T cell abundances (Fig. 6A). Additionally, we explored the associations of KAT2A expression across immune and molecular subtypes (Fig. 7A). The top 5 KAT2A expression was PRAD, TGCT, STAD, BRCA, and LIHC. Then, we focused on these five cancer and assessed the expression of six immune and molecular subtypes. The main clinical and molecular characteristics were depicted as C1 (wound healing); C2 (IFN-gamma dominant); C3 (inflammatory); C4 (lymphocyte depleted); C5 (immunologically quiet); C6 (TGF-b dominant). KAT2A gene expression was similar in BRCA (Fig. 7B), PRAD (Fig. 7D), and STAD (Fig. 7E) within five immune and molecular subtypes. KAT2A expression decreased in C6 (LIHC) and C3/C4 (TGCT) in LIHC (Fig. 7C) and TGCT (Fig. 7F).
Associations between KAT2A expression and immune subtypes across human cancers. A. Associations of KAT2A expression across immune cells in the TISIDB database. B–F. Correlation of KAT2A expression in BRCA, LIHC, PRAD, STAD, and TGCT within immune subtypes. C1 (wound healing); C2 (IFN-gamma dominant); C3 (inflammatory); C4 (lymphocyte depleted); C5 (immunologically quiet); C6 (TGF-b dominant). 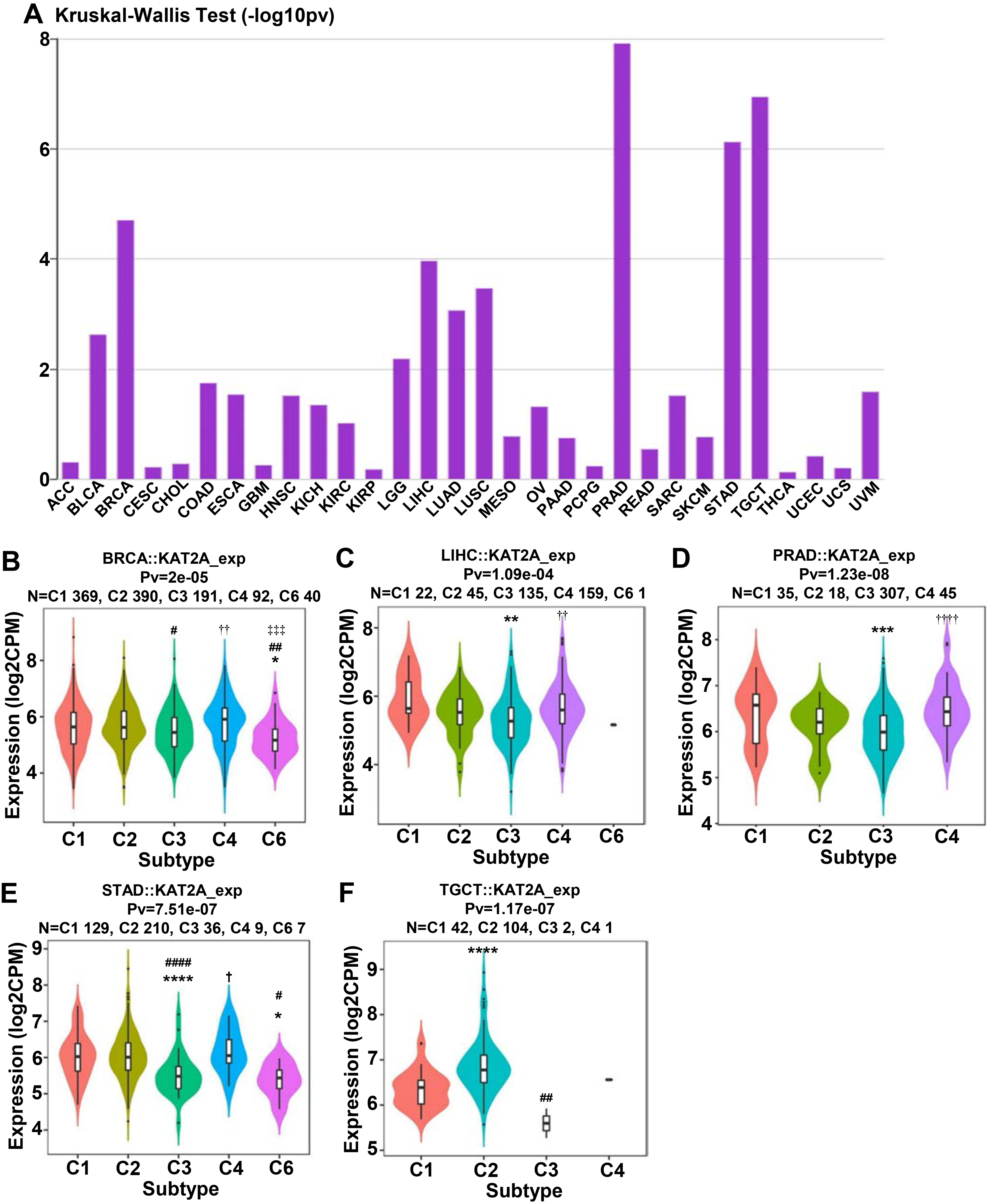
KAT2A gene expression level and overall survival. A. Associations of overall survival with KAT2A expression across human cancers. B–E. Kaplan–Meier survival curves comparing high and low KAT2A expression in KIRC, PAAD, STAD, and LGG. High KAT2A expression was related to unfavorable OS in KIRC and better survival in PAAD, STAD, and LGG. Thus, KIRC patients with high KAT2A expression have a risk factor affecting survival.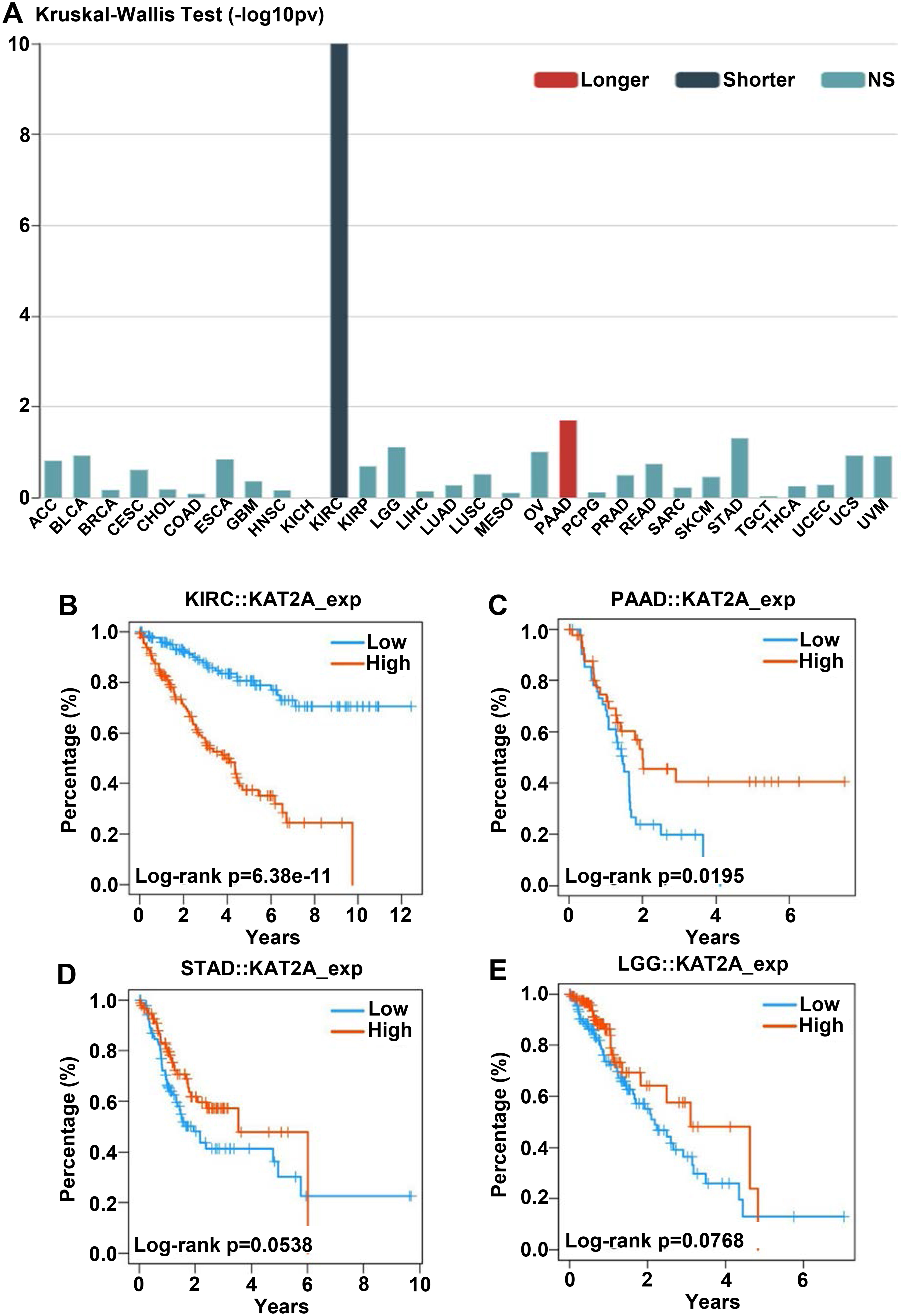
Furthermore, we assessed the associations of overall survival with KAT2A expression across human cancers. We determined the relationship between longer, shorter, and NS (no significant) overall survival rates with KAT2A expression among 30 cancer (Fig. 8A). Shorter overall survival rate was significantly correlated with elevated KAT2A expression in KIRC. Low KAT2A expression was significantly correlated with a longer overall survival rate in PAAD (Fig. 8A). We also analyzed the connections between overall survival rate and KAT2A expression in KIRC, PAAD, STAD, and LGG using Kaplan-Meier curves. A higher KAT2A expression indicates a shorter survival rate within 12 years (
KAT2A expression in human normal and tumor tissues
Dysregulation of transcriptomes is closely associated with cancer, leading to alterations in specific genes or pathways that contribute to uncontrolled tumor growth [49]. Over 500 genes have been identified as strongly implicated in transforming normal cells into cancer cells [50]. However, tracking total mRNA levels is insufficient for the major feature of KAT2A expression and cancers. Translation and (or) translocation of mRNAs encoding proteins that govern cancer cell plasticity play a central role in tumor cell differentiation [51, 52]. Thus, to provide a comprehensive view of KAT2A expression and cancers, we examined the KAT2A protein and mRNA level patterns in human cancer tissues. KAT2A protein was mainly located intracellularly and expressed with low tissue specificity. To validate the KAT2A expression in these organs, we used colon, spinal cord, kidney, liver, pancreas, and stomach tumors and normal human tissue sections to analyze pathological KAT2A levels with IHC staining. In the tumor cores, colon, spinal cord, kidney, liver, pancreas, and stomach cancer cells and tumor stroma were positive for KAT2A (Fig. 9A, lower panel). Different tumor organs demonstrated a range of KAT2A intensity. The normal human colon, spinal cord, kidney, liver, pancreas, and stomach organs were also positive for KAT2A (Fig. 9A, upper panel). Next, we evaluated KAT2A levels among these organs. KAT2A mRNA levels greatly increased in tumor organs compared to normal ones by qPCR (Fig. 9B), 2-4 folds compared to their respective control tissues. Finally, we performed a Western blot assay with KAT2A and GAPDH antibodies and observed higher KAT2A protein levels in tumors than their respective control organs (Fig. 9C). Accordingly, the quantification data showed a similar higher KAT2A protein level using the GAPDH housekeeping gene for normalization (Fig. 9D). Therefore, KAT2A expression in tumors presented a comprehensive range compared to normal samples, consistent with the database analysis (Fig. 1).
KAT2A expression in normal and cancer tissues. A. Representative immunohistochemistry (IHC) staining of KAT2A in human normal and tumor colon, spinal cord, kidney, liver, pancreas, and stomach tissues with KAT2A antibody. Scale bar 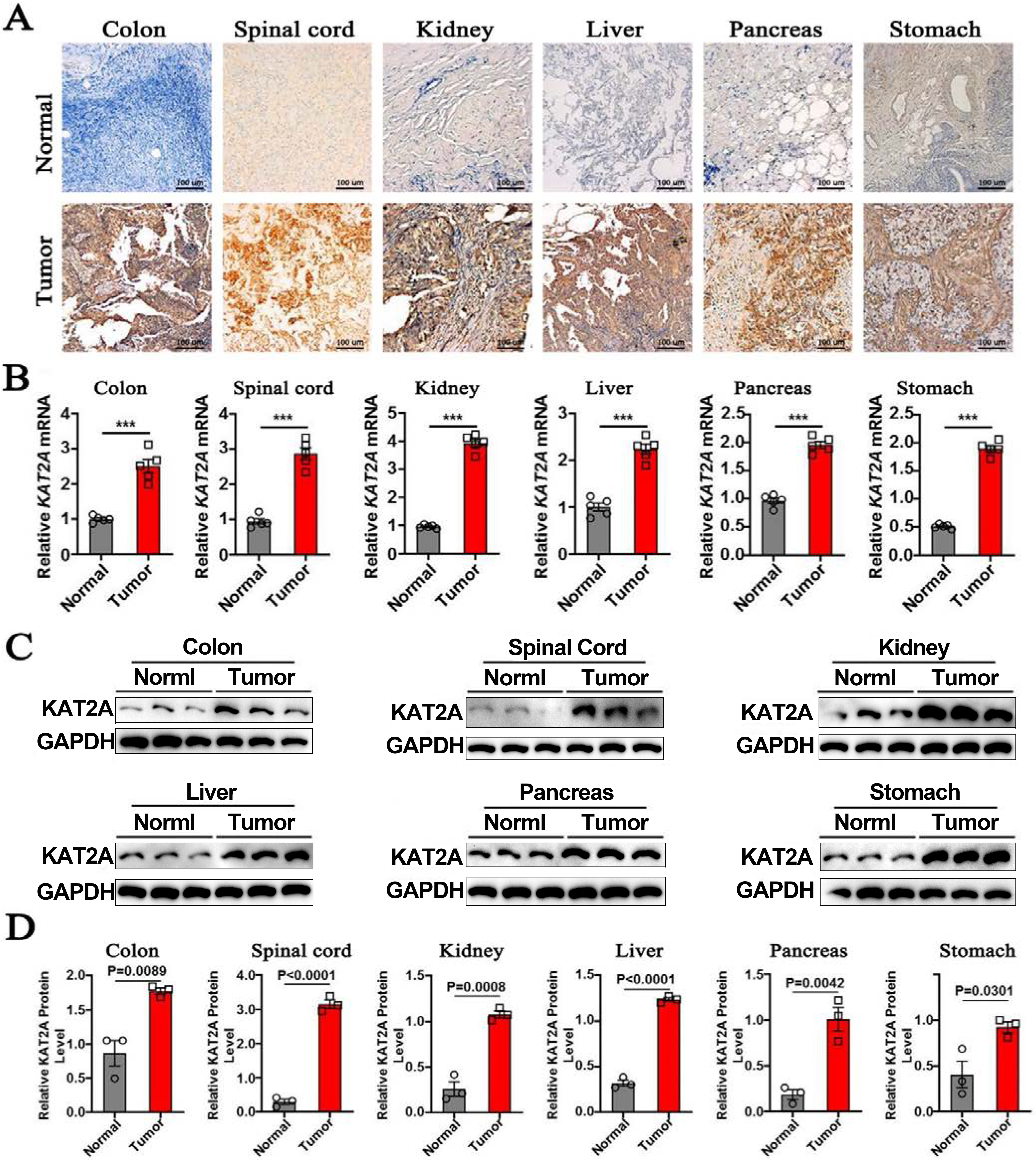
Pathologically, dysregulation of epigenetic events can lead to cancer. Consequently, identifying drugs that can modify these epigenetic changes holds great clinical potential. As described, KAT2A-mediated lysine acetylation is a reversible post-translational modification involved in tumor genesis [53]. Small molecules or inhibitors that can modulate the HAT or its competence, such as HDACs (histone deacetylases), can be potential therapeutics for various diseases [54, 55]. Hence, to explore the potential mechanisms in which KAT2A participates in carcinogenesis, we used the TISIDB online tool to predict the drugs targeting KAT2A collected from the DrugBank database (Fig. 10A. DB01992 has a direct interaction with KAT2A and is targeted by other modules. DB01992 is a small molecule of Coenzyme A (Fig. 10B) known for its role in the synthesis and oxidation of fatty acids and pyruvate in the citric acid cycle. Furthermore, we searched the HISTome2 database for more information on HAT inhibitors. Gallic acid, Garcinol, Anacardic acid, Procyanidin, MB-3, CTK7A, Plumbagin, and Embelin were predicted to interact with KAT2A directly (Fig. 10C-J) [56, 57]. Overall, these results might accelerate drug understanding and future development.
Drug targeting KAT2A in the DrugBank database. Prediction of drugs targeting KAT2A in the DrugBank database. B. Structure of Coenzyme A DB01992. C–J. Structures of HAT inhibitors: Gallic acid (C), Garcinol (D), Anacardic acid (E), Procyanidin (F), MB-3 (G), CTK7A (H), Plumbagin (I), and Embelin (J) from the HISTome2 database.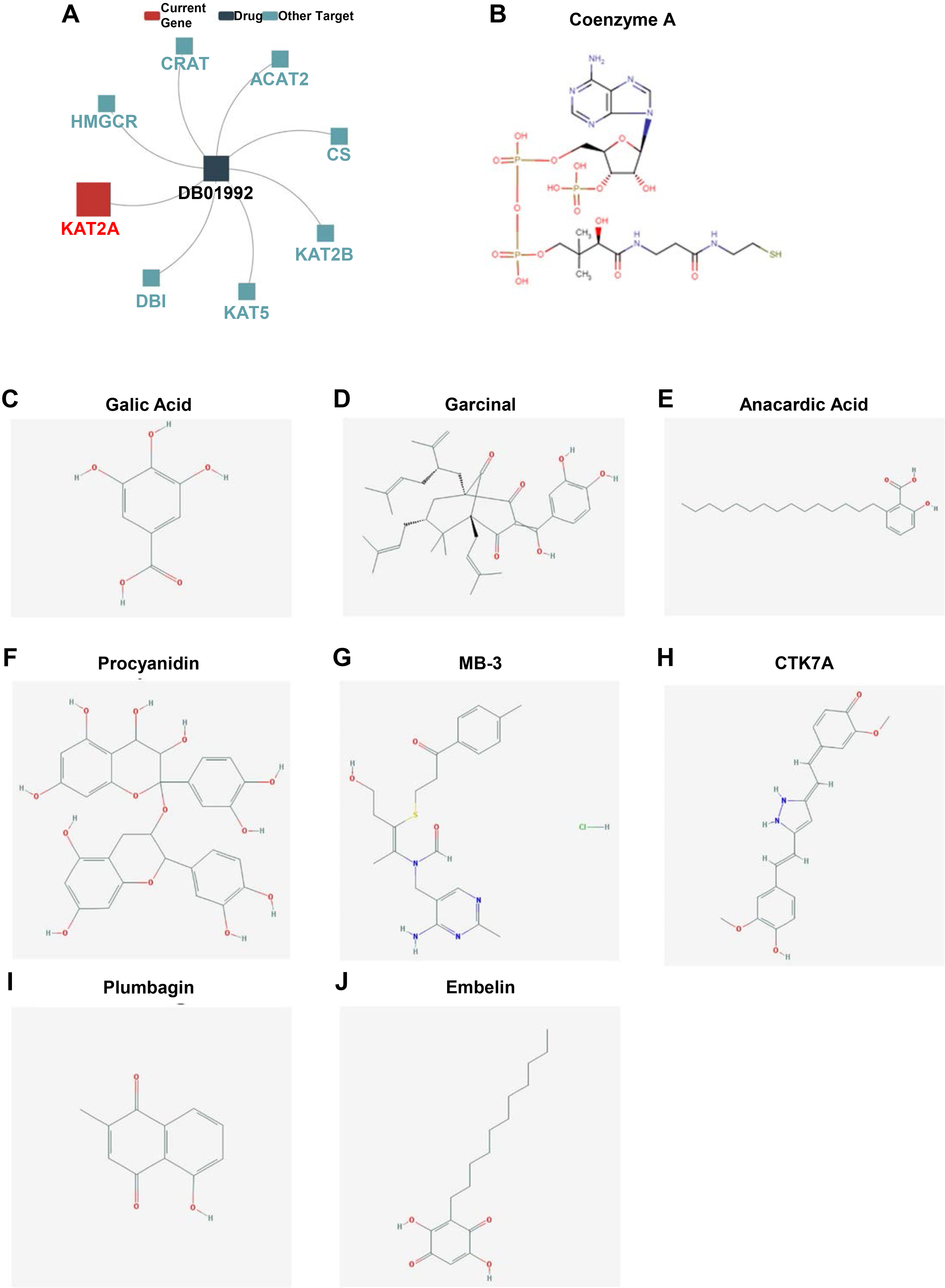
Aberrant epigenetic modifications are closely associated with the risk of numerous human cancers. Changes in the epigenome have significant consequences on the regulation of tumorigenesis, invasiveness, and metastasis. Emerging evidence suggests that KAT2A is differentially expressed in metabolic disorders and is critical in multiple human cancers. However, the mechanism by which the histone-modifying complex rewrites and influences global epigenome levels in pan-cancer remains incomplete.
Herein, we conducted a pan-cancer analysis of KAT2A expression profiles encompassing 33 tumor types. We explored the role of KAT2A, including its expression profile, sexually dimorphic characteristics, correlation with pathological stages, overall survival analysis, immune landscape, and potential drugs. Notably, KAT2A expression was upregulated in seven cancer types: BRCA, KIRC, LIHC, LUAC, LUSC, PRAD, and THCA (Fig. 1). However, we did not found sex difference patterns in normal or cancer samples, indicating that sexually dimorphic patterns in most cancer types are not caused by KAT2A (Fig. 2). Since high KAT2A levels were correlated with elevated tumor risk, we evaluated whether KAT2A expression was correlated with poor patient prognosis in different cancer stages. KAT2A expression was significantly associated with tumor stage in BLCA, HNSC, KICH, KIRP, LIHC, LUSC, and READ cancer samples (Fig. 3), which is partially consistent with the conclusion from Fig. 1; for example, LIHC and LUSC showed increased KAT2A levels, KAT2A upregulation was positively correlated with unfavorable prognosis, and KAT2A was correlated with cancer infiltration levels in LIHC and LUSC.
Genes seldom operate in isolation within cellular processes, while KAT2A is a subunit of two distinct macromolecular complexes (SAGA and ATAC) that drive histone acetylation in the promoter region of target genes. To understand the properties and functions of the KAT2A gene, we used the STRING database to estimate regulatory relationships between KAT2A and its interacting partners based on Pearson’s correlation coefficient. KAT2A was positively correlated with TADA3, CCDC101, TRRAP, SUPT3H, MYC, TADA2A, and USP22, while negatively correlated with TADA2B and ATXN7 (Fig. 4). Additionally, we explored KAT2A gene alterations that are associated with various genomic mutations across cancers. Notably, mutations were among the most significant single factors for alteration in UCEC and SKCM. Also, KAT2A amplification frequencies were elevated in ESCA, STAD, PAAD, MESO, and PCPD (Fig. 5), indicating that various genetic alterations were assembled in KAT2A that led to cancer development. One relevant pathway of KAT2A gene rewiring and epigenetic landscape is histone acetylation, which plays a critical role in regulating chromatin structure and participating in specific gene regulation, affecting the development and progression of diverse cancers. We also investigated the correlation between KAT2A and immune infiltration, as well as immune checkpoints, using the TIMER database. KAT2A expression was negatively correlated with the abundance of active CD8
Generally, KAT2A contributes to cancer development via transcriptional activity control. Namely, KAT2A regulates E2F and MYC transcriptional targets through histone acetylation-mediated co-activation. Enhanced KAT2A is associated with a bad prognosis in breast cancer [58, 59], Non-Small Cell Lung Carcinoma [60, 61], and Colon [15, 62]. Recently, the nonhistone substrates of KAT2A have been reported in cancer biology, as well as their potential roles in the development and progression of cancer [63, 64]. KAT2A can regulate the activity of Peroxisome Proliferator-Activated Receptor Gamma-Coactivator-1
However, although we investigated and integrated KAT2A’s role in these cancers with different databases and bioinformatics analyses, our study also has some limitations. First, it is a reanalysis study with the public data; only modern bioinformatics methods were employed to explore the relations. We have few original data to reproduce the results with the new criteria and algorithm, and further validation might support our conclusions. Second, due to the small number of samples, we could not distinguish KAT2A expression patterns between groups for the human sample’s validation data. Thus, this result is not comprehensive without sufficient sample size and clarification by pharmacodynamic and molecular biological experiments data. Moreover, KAT2A-mediated histone acetylation is only one part of the epigenetic modification. The intracellular signal transduction and regulatory factor activity are complex and fluctuate irregularly, and it was still unclear how KAT2A affected clinical survival via the immune pathway.
Conclusion
In summary, we explored the correlation and the prognostic significance of KAT2A expression in diverse human cancers. KAT2A was a predictive cancer factor upregulated in BRCA, KIRC, LIHC, LUAD, LUSC, PRAD, and THCA and correlated with prognosis in KIRC, PAAD, STAD, and LGG. Therefore, we shed some light on a better understanding of the underlying molecular mechanisms and crucial molecular players of KAT2A and provide a new viewpoint for researchers to target the causes of cancers.
Abbreviation list
Ethics approval and consent to participate
This study was approved by the clinical trial ethical committee of the Shanghai Pulmonary Hospital, Tongji University School of Medicine. After approval of the ethical committee, all methods and information collection in this study were performed according to the relevant guidelines and regulations.
Consent for publication
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the authors at reasonable request. The code used throughout this study is available upon reasonable request from the corresponding authors.
The datasets presented in this study can be found in the following databases: UCSC Xena (
Competing interests
The authors declare that they have no competing interests.
Funding
None.
Authors’ contributions
Conception: Lu-zong Yang and Ji Liu.
Interpretation or analysis of data: Hua Li, Chun Li, and Ji Liu.
Preparation of the manuscript: Chun Li, Ji Liu, and Hua Li.
Revision for important intellectual content: Ji Liu and Hua Li.
Supervision: Ji Liu.
All authors read and approved the final version of the manuscript.
Footnotes
Acknowledgments
Not applicable.