
Abstract
Emerging evidence has revealed a relationship between lamin B1 (LMNB1) and several cancers such as cervical cancer, liver cancer, and prostate cancer. But no systematic pan-cancer analysis is available. Little is known about the clinical significance and biomarker utility of LMNB1. In this study, we first revealed the key role of LMNB1 in esophageal carcinoma (ESCA) through weighted gene co-expression network analysis (WGCNA) and disease-free survival (DFS) analysis. Based on this result and the datasets of the cancer genome atlas (TCGA), we explored the biomarker utility of LMNB1 across thirty-three tumors. We found that LMNB1 was highly expressed in most of the cancers and significant associations existed between LMNB1 expression and prognosis of cases of nearly half of the cancers. We also found that LMNB1 expression was associated with the infiltration level of Macrophages M1 and T cells CD4 memory activated in some cancers. Moreover, LMNB1 was mainly involved in the functional mechanisms of MRNA binding, olfactory transduction, and gene silencing. Our study first provides a pan-cancer study of LMNB1, thereby offering a relatively comprehensive understanding of the biomarker utility of LMNB1 across thirty-three tumors.
Introduction
Cancer has biological characteristics such as abnormal cell proliferation, differentiation, invasiveness, and metastasis [1]. Therefore, effecting a radical cure for most cancers remains the enduring millstone worldwide [2]. ESCA is one of the most aggressive cancers, thereby easily making negative patients develop systemic relapse [3]. ESCA ranked seventh in terms of incidence (32,000/1000,000 signifying that ESCA was responsible for an estimated 32 in every 1000 new cancer cases) and sixth in mortality overall (53000/1000,000 signifying that ESCA was responsible for an estimated 53 in every 1000 cancer deaths) in 2018 [2, 4]. As manifested by Torre et al, increased obesity and heavy drinking and smoking are the major risk factors for ESCA [5]. However, in fact, the underlying causes of driving the neoplastic process are often associated with oncogene and tumor-suppressor gene mutations [6]. Therefore, there is an urgent need to explore novel cancer biomarkers, thereby promoting the development of cancer diagnosis and treatment.
LMNB1 is a component of the nuclear lamina. It is known as a protein coding gene which encodes one of the two B-type lamin proteins [7]. LMNB1 functions in nuclear envelope lamina and possesses a transcriptional coregulatory activity having an important role in DNA replication, cellular aging, and stress responses [8, 9, 10]. Clinical investigations have indicated that LMNB1 which cooperates with vimentin may be an early symptomatic marker for liver tumor [11, 12]. LMNB1 overexpression is also closely related to the poor differentiation of pancreatic cancer [13]. And the expression of LMNB1 suggests the adverse prognosis of the breast cancer patients [14]. However, there is still no pan-cancer study to illustrate clinical significance and biomarker utility of LMNB1 in cancers based on big clinical data.
In this study, firstly, we found the key role of LMNB1 in ESCA through WGCNA and DFS analysis. Subsequently, we conducted a systematic pan-cancer analysis of LMNB1 to elucidate its biomarker utility in 33 cancer types. Its pan-cancer analysis was based on a group of factors including gene expression, survival status, tumor mutation burden (TMB) and microsatellite instability (MSI) scores, immune infiltration, and gene enrichment.
Materials and methods
Datasets and pre-processing
In this study, 33 cancer types were considered including adrenocortical carcinoma (ACC), bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma (CESC), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), lymphoid neoplasm diffuse large B cell lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), brain lower grade glioma (LGG), head and neck squamous cell carcinoma (HNSC), kidney chromophobe (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), acute myeloid leukemia (LAML), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), mesothelioma (MESO), ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), sarcoma (SARC), skin cutaneous melanoma (SKCM) stomach adenocarcinoma (STAD), testicular germ cell tumors (TGCT), thyroid carcinoma (THCA), thymoma (THYM), uterine corpus endometrial carcinoma (UCEC), uterine carcinosarcoma (UCS), and uveal melanoma (UVM). The gene expression RNAseq (HTSep – FPKM), clinical survival data and somatic mutation (SNPs and small INDELs) of 33 cancers were extracted from UCSC Xena (
Construction of the gene co-expression network and identification of clinically significant modules
The WGCNA package [15] in R software is a tool to construct a gene co-expression network. In the expression data of ESCA, the top 25% genes with the largest variance were selected to be submitted to WGCNA package for constructing a gene co-expression network. Then, WGCNA package was applied to calculate the correlation between genes and weight to make the whole network approximate the scale-free network distribution. In this process, a suitable soft threshold was selected when R
After obtaining the modules, to identify the clinically significant modules, we used “module eigenvalues” to represent the combined value of the gene set expression of the module. Meanwhile, ESCA and its control samples were selected as the clinical phenotypes for subsequent correlation analysis. Therefore, each module could be associated with ESCA according to the phenotype correlation coefficient and the saliency
Identification of hub genes in key modules and DFS analysis
Hub gene is a highly connected gene in the co-expression network module. The correlations between genes and sample traits were calculated as gene significance (GS). The correlations between the module’s own genes and gene expression profiles were defined as module membership (MM). After screening the key gene modules associated with ESCA, we calculated GS for ESCA and MM for each gene in the key modules and drew a scatterplot of GS vs MM for each key module. The genes with GS
Pan-cancer analysis of LMNB1
Pan-cancer analysis aims to examine similarities and differences between genome and cellular changes in different cancer types [16, 17]. In this study, pan-cancer analysis of LMNB1 mainly included analyses of gene expression, survival status, TMB and MSI scores, immune infiltration and gene enrichment.
Gene expression analysis
In this study, firstly, the expression matrix of LMNB1 in 33 cancers was extracted and integrated by perl software. Based on the expression matrix of LMNB1, wilcoxon test was used to test the expression difference of LMNB1 between tumor tissues and the corresponding normal tissues in the 24 cancers with both tumor samples and normal samples. Finally, we used the ggpubr R package to visualize the results of difference expression of LMNB1 in these cancers. Considering that there were 9 cancers with no normal samples, we also evaluated the expression level of LMNB1 by GEPIA database which combines the data for normal tissues from the GTEx database with the data from TCGA [18], and 31 cancers were tested. Additionally, we used the Kruskal-Wallis test to assess the expression difference of LMNB1 between different pathological stages for 21 tumors with corresponding information of pathological stages.
Workflow chart of our study.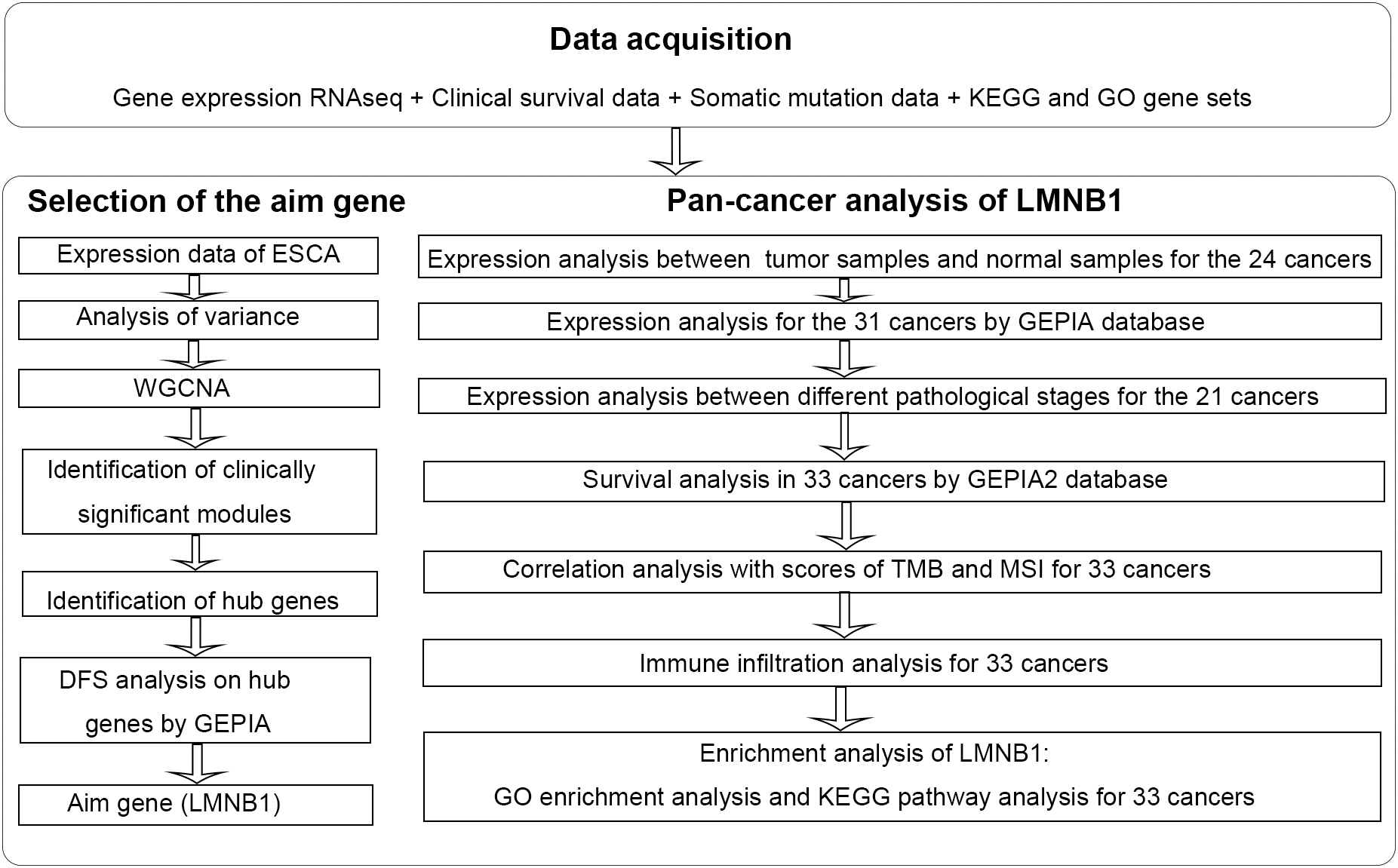
The relationship between LMNB1 expression and clinical outcome including overall survival (OS) and DFS was identified in GEPIA2 online analysis data-base [19]. OS refers to the period between initial diagnosis and death date (due to any cause) [20]. DFS is defined as the length of time between the start of randomization and the recurrence of the disease or death (for any reason) [21]. The “Survival Map” function of GEPIA2 was used to obtain the OS and DFS significance maps of LMNB1 across 33 tumors. And the “Survival Analysis” function of GEPIA2 was used to obtain the OS and DFS survival plots. The expression thresholds for splitting the high-expression and low-expression cohorts were set as Cutoff-high (50%) and cutoff-low (50%) values, and the log-rank test was applied in the hypothesis test.
Correlation analysis with scores of TMB and MSI
TMB and MSI were effective prognostic biomarkers and immune therapy response indicators in many tumors. TMB was defined as the total number of somatic gene coding errors, gene insertion and deletion errors, and base substitutions detected per million bases. And MSI was defined as total incidence of deletion or insertion in repeating sequences per million base pair. In this study, the associations between LMNB1 and scores of TMB and MSI were analyzed for the 33 cancers using the Spearman’s rank correlation test in R software.
Immune infiltration analysis
Immune cells infiltrating tumors can profoundly affect tumor progression and the success of anti-cancer treatments. In this work, we investigated the potential relationship between LMNB1 expression and the infiltration level of the immune cells of T cells CD4 memory activated and Macrophages M1 in 33 cancer types. The CIBERSORT algorithm was applied for immune infiltration estimations. The P-values and partial correlation values were received via the Spearman’s rank correlation test. The analysis results of
Enrichment analysis of LMNB1
We divided the samples into high and low expression groups according to the expression median of LMNB1. Based on the KEGG and GO gene sets, we used the clusterProfiler [22] package in R software to perform KEGG pathway analysis and GO enrichment analysis for the 33 cancers.
Results
Identification of clinically significant modules by WGCNA
14130 genes in the top 25% largest variance were used to construct the weighted gene co-expression network. Firstly, a sample clustering tree was constructed (Fig. 2A). We did not find obvious outlier samples. Secondly, as shown in Fig. 2B and C, the soft threshold reached 4 when
Construction of WGCNA co-expression modules for the genes with top 25% largest variance. (A) A sample clustering tree to detect obviously outliers. (B) Analysis of the scale-free fit index for various soft threshold to determine soft threshold in WGCNA. (C) The mean connectivity for various soft threshold to determine soft threshold in WGCNA. (D) Dendrogram of genes clustered based on the Dynamic Tree Cut to obtain the modules. (E) Eigengene network heatmap which summarizes the modules yielded in the clustering analysis.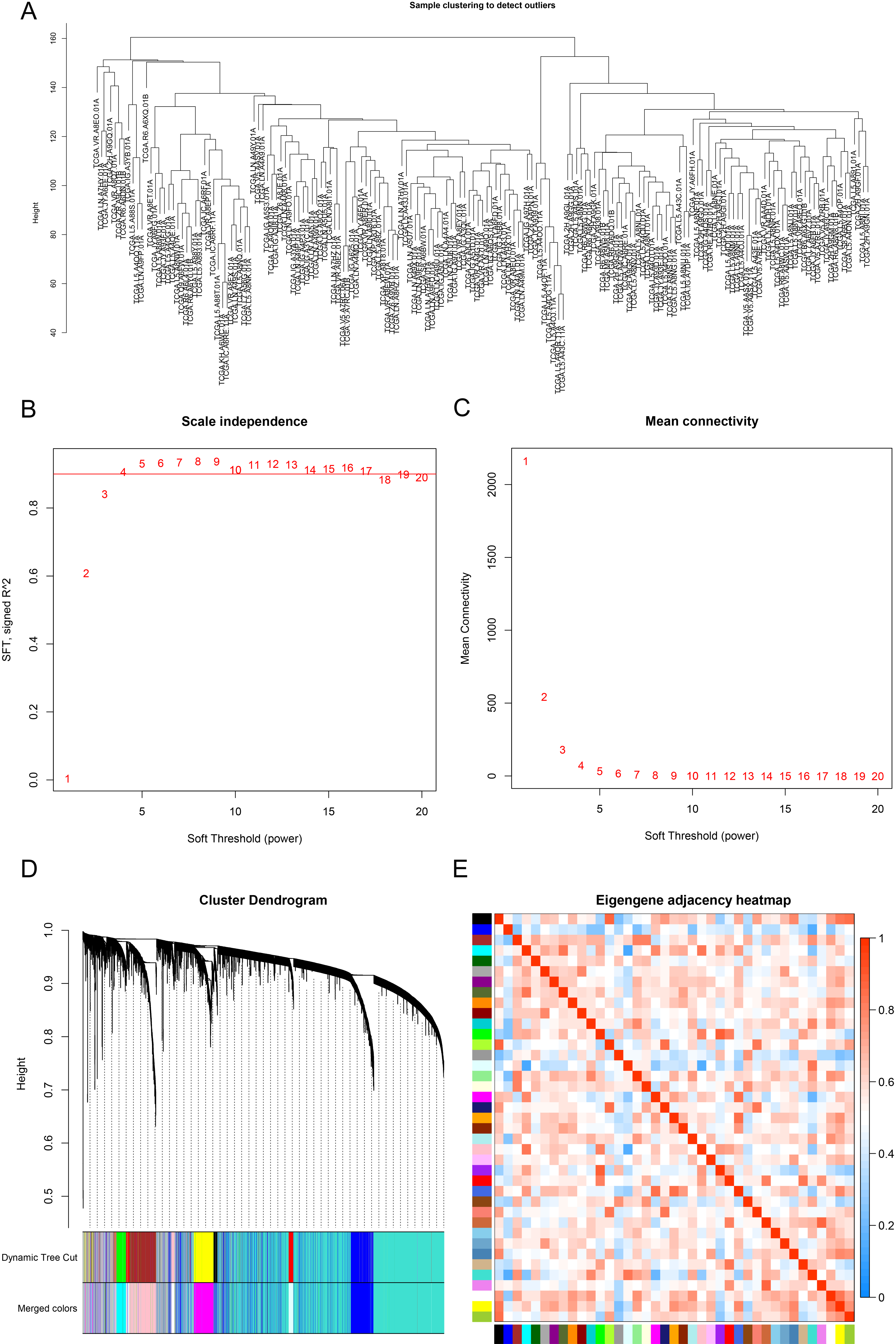
Heat map of the correlation between module eigengenes and clinical traits where control represents normal tissues and ESCA represents tumor tissues. The phenotype correlation coefficients (the decimals) and 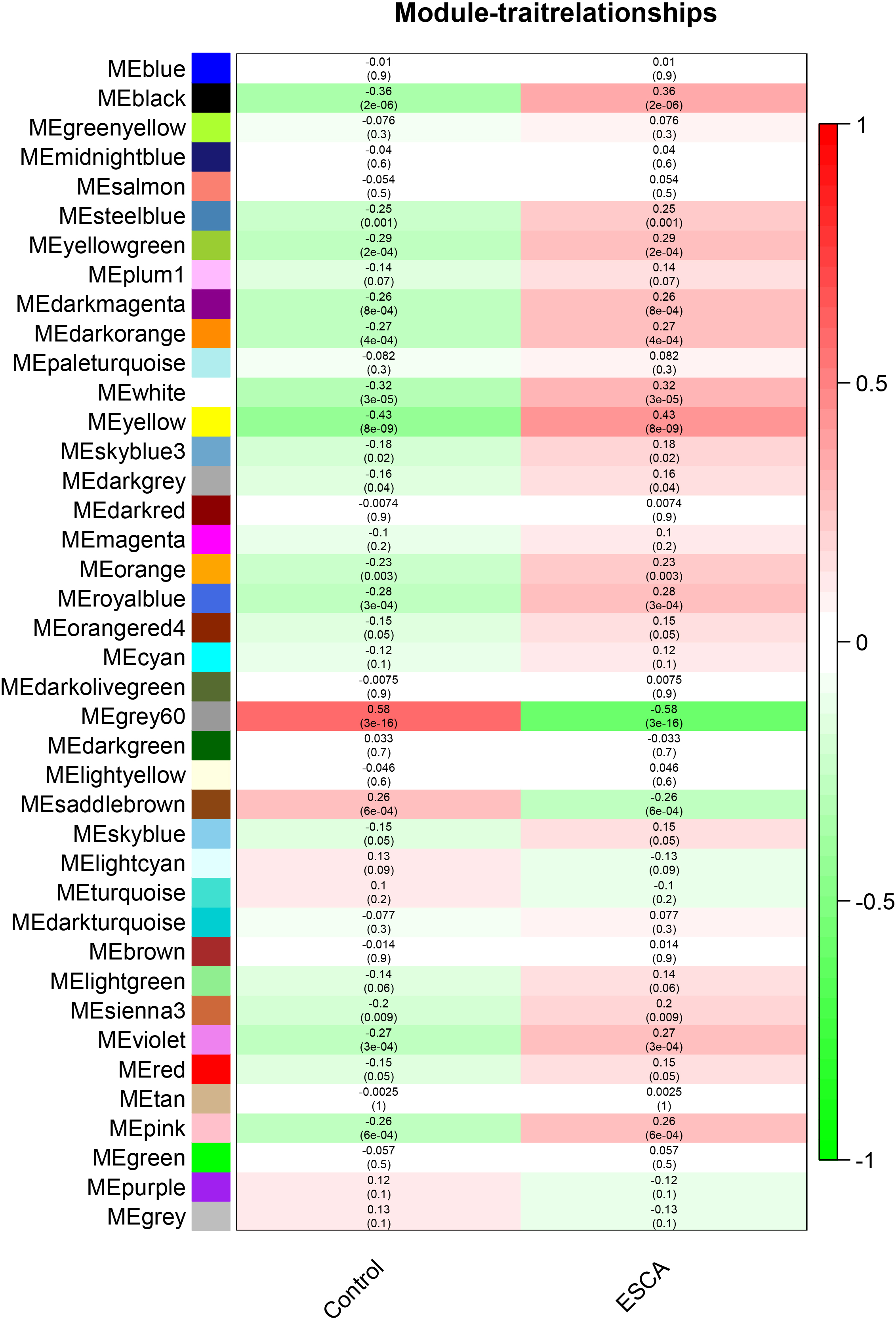
After computing GS for ESCA and MM for each gene, 92 genes in the two key modules were considered as hub genes by the thresholds of GS
Acquisition of hub genes and DFS analysis in ESCA. (A) GS vs MM in yellow module to select hub genes (the purple area). (B) GS vs MM in grey60 module to select hub genes (the purple area). (C) Correlation between LMNB1 expression and survival prognosis of ESCA patients.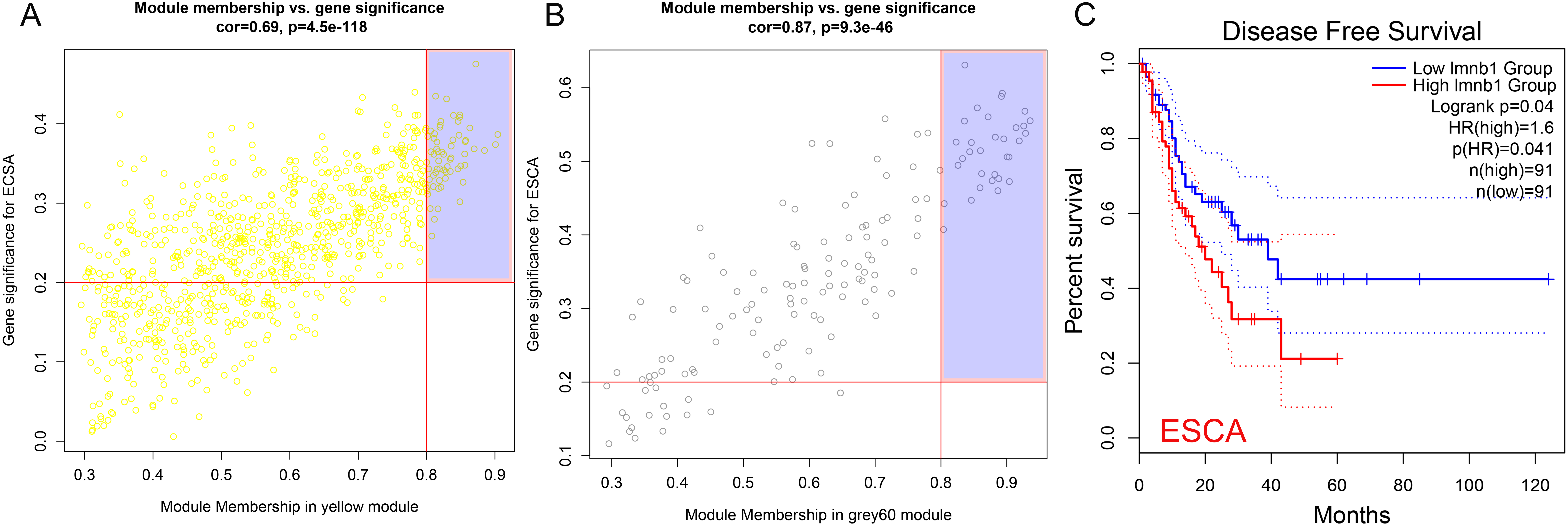
The expression levels of LMNB1 in different cancers. Red (blue) cancer names indicate high (low) expression of LMNB1 compared with the corresponding normal tissues. 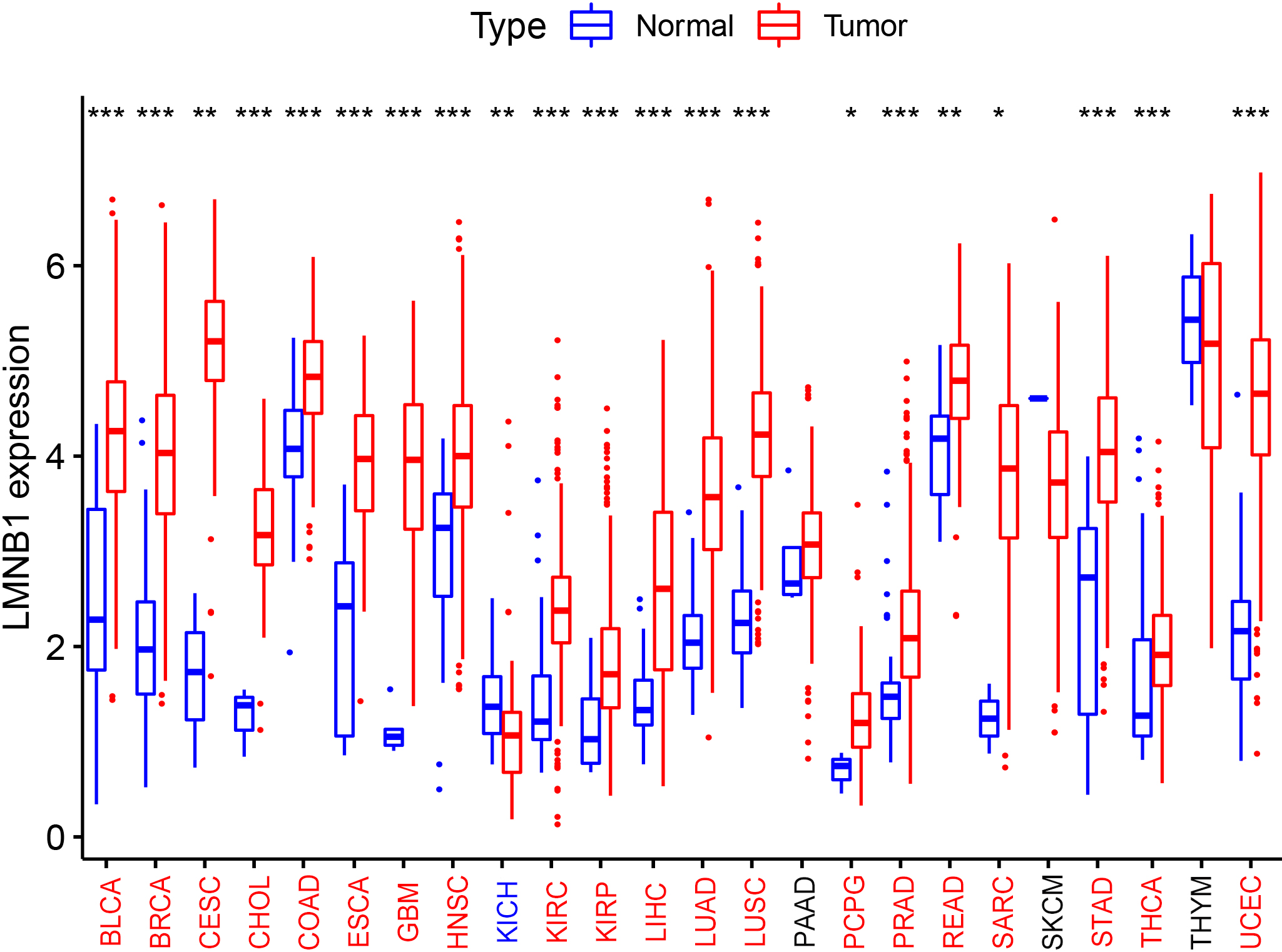
The expression levels of LMNB1 in pan-cancer
We analyzed the expression of LMNB1 between tumor tissues and the corresponding normal tissues across 24 cancer types of TCGA. As shown in Fig. 5, the expression levels of LMNB1 were significantly higher in tumor tissues (
The expression levels of LMNB1 in 31 cancers from GEPIA database. Red cancer names indicate high expression of LMNB1 compared with the corresponding normal tissues. Num(T) represents the number of tumor samples, and num(N) represents the number of normal samples. 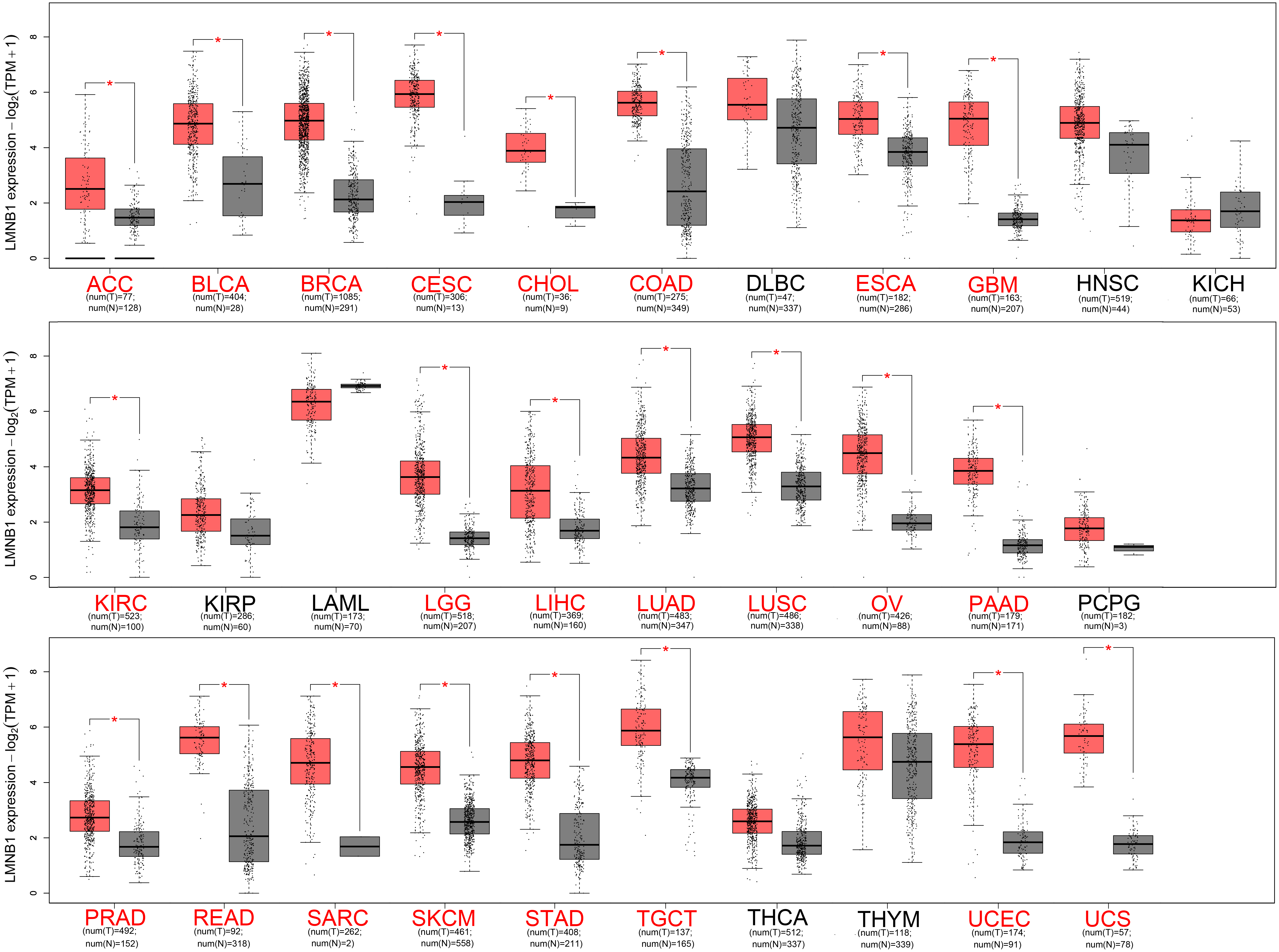
Further, from the perspective of total P value, we observed that the expression of LMNB1 was significantly different between the different pathological stages (
The expression levels of LMNB1 are analyzed for the 21 tumors with corresponding information of pathological stage. For the box plot of each cancer, only 11 box plots with total 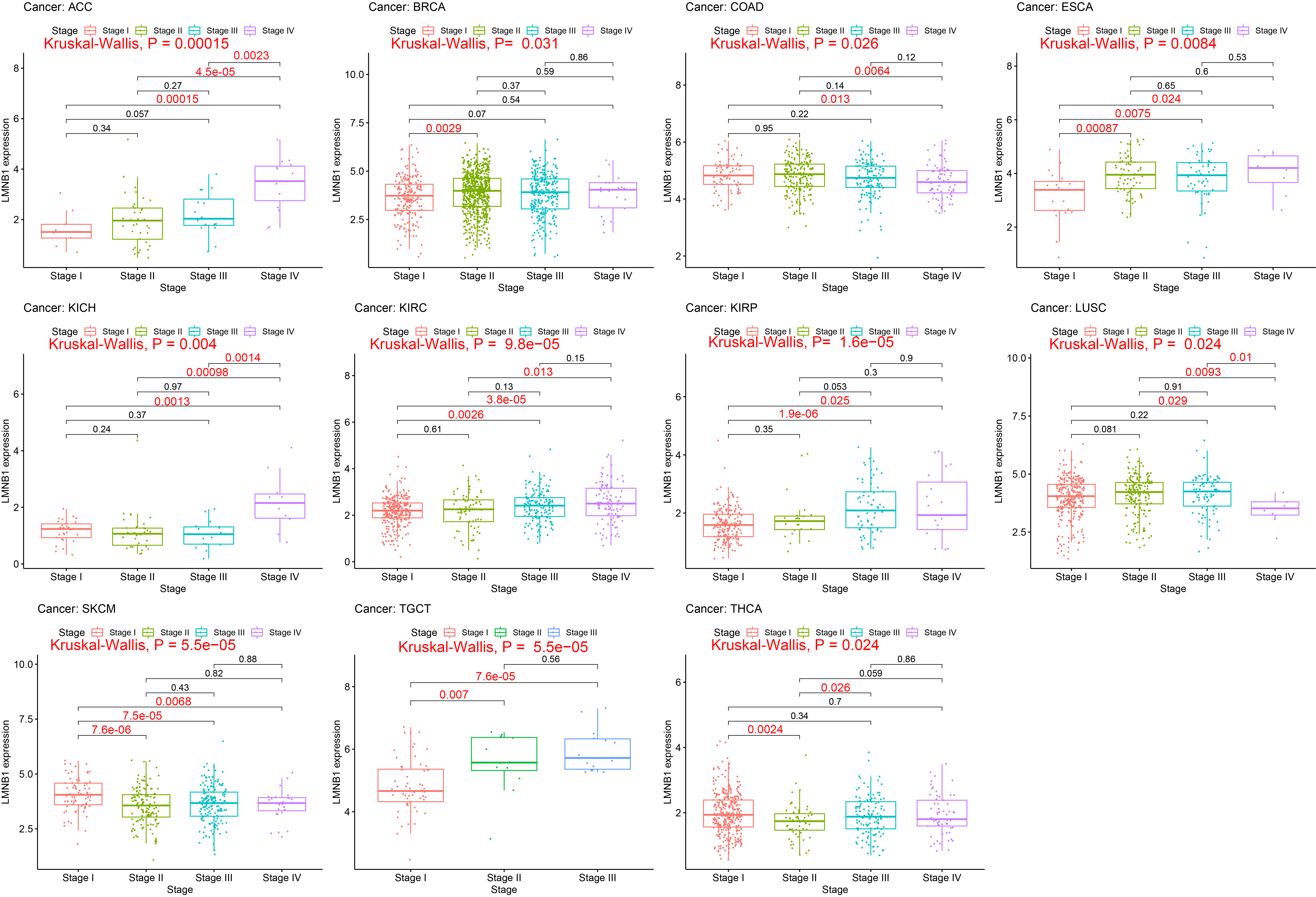
We investigated the correlation of LMNB1 expression and the prognosis of patients in different tumors. As shown in Fig. 8A, for the OS, high expression of LMNB1 was linked to poor prognosis of 9 types of cancer including ACC (
Correlation between LMNB1 expression and survival prognosis of the 33 cancers in TCGA. The survival maps (small colored boxes) were heat map of HR, in which red (blue) color represented that LMNB1 played a detrimental (protective) prognostic role in the corresponding cancer. HR 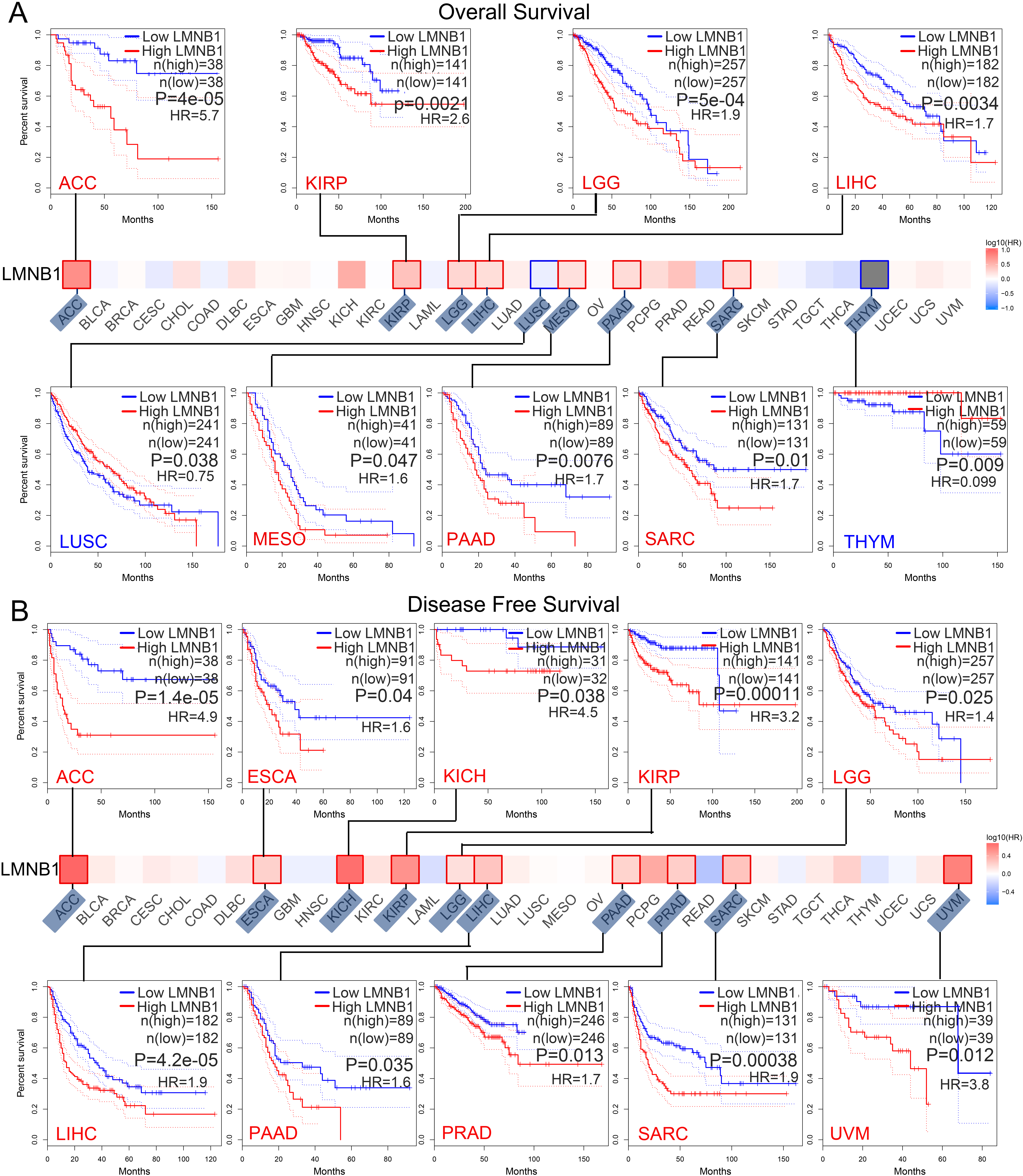
The correlation between LMNB1 expression and scores of TMB and MSI was evaluated (Table 1). As shown in Fig. 9A, the correlation between LMNB1 expression and TMB achieved significance (
Correlation analysis of LMNB1 expression with TMB and MSI
Correlation analysis of LMNB1 expression with TMB and MSI
Relationship between LMNB1 expression levels and TMB and MSI in all the 33 cancer types in TCGA database by spearman correlation test. (A) Correlation between TMB and LMNB1 expression. (B) Correlation between MSI and LMNB1 expression. 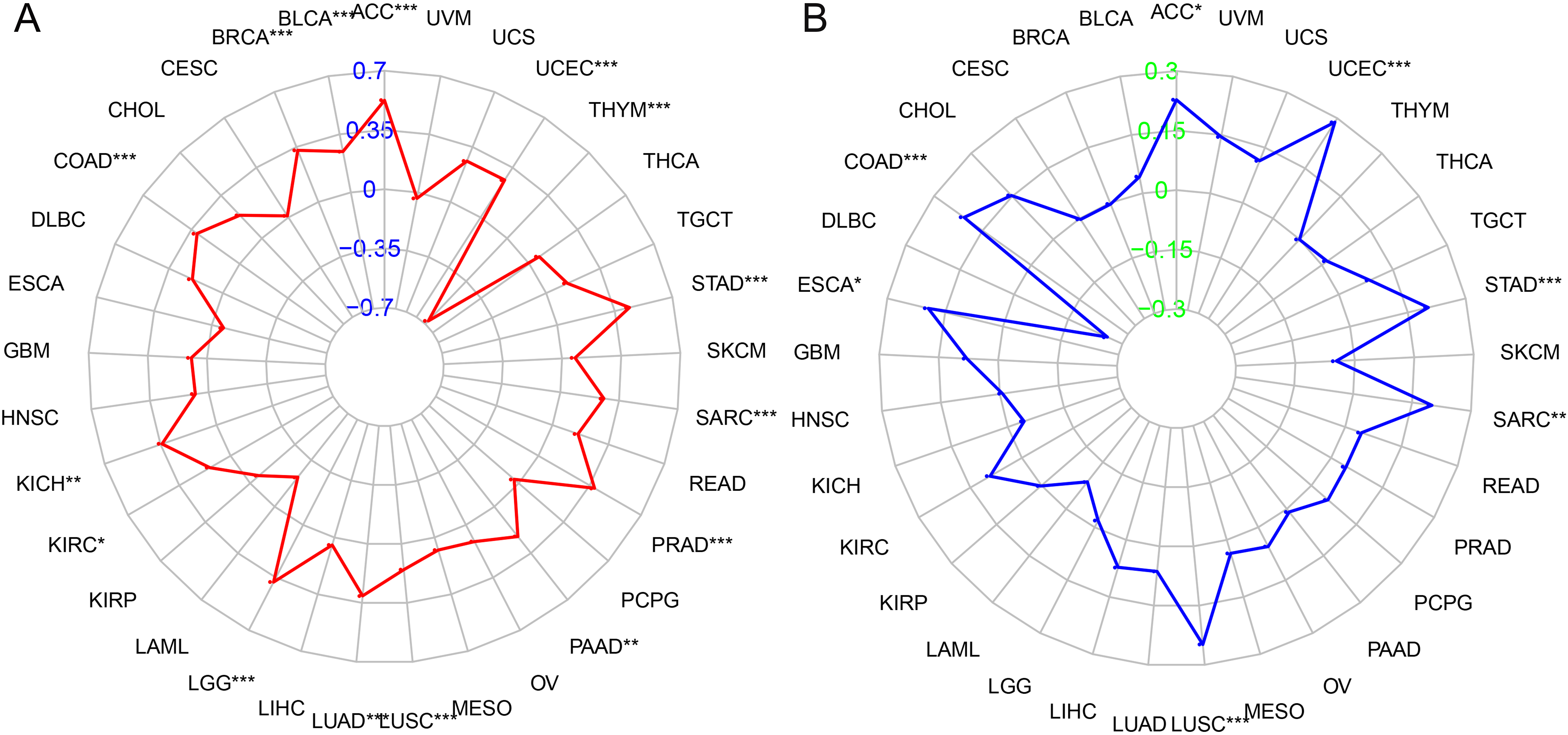
In the immune infiltration analysis, we observed a statistically positive correlation (
Relationship between the infiltration level of Macrophages M1 and LMNB1 expression. The correlation coefficients and 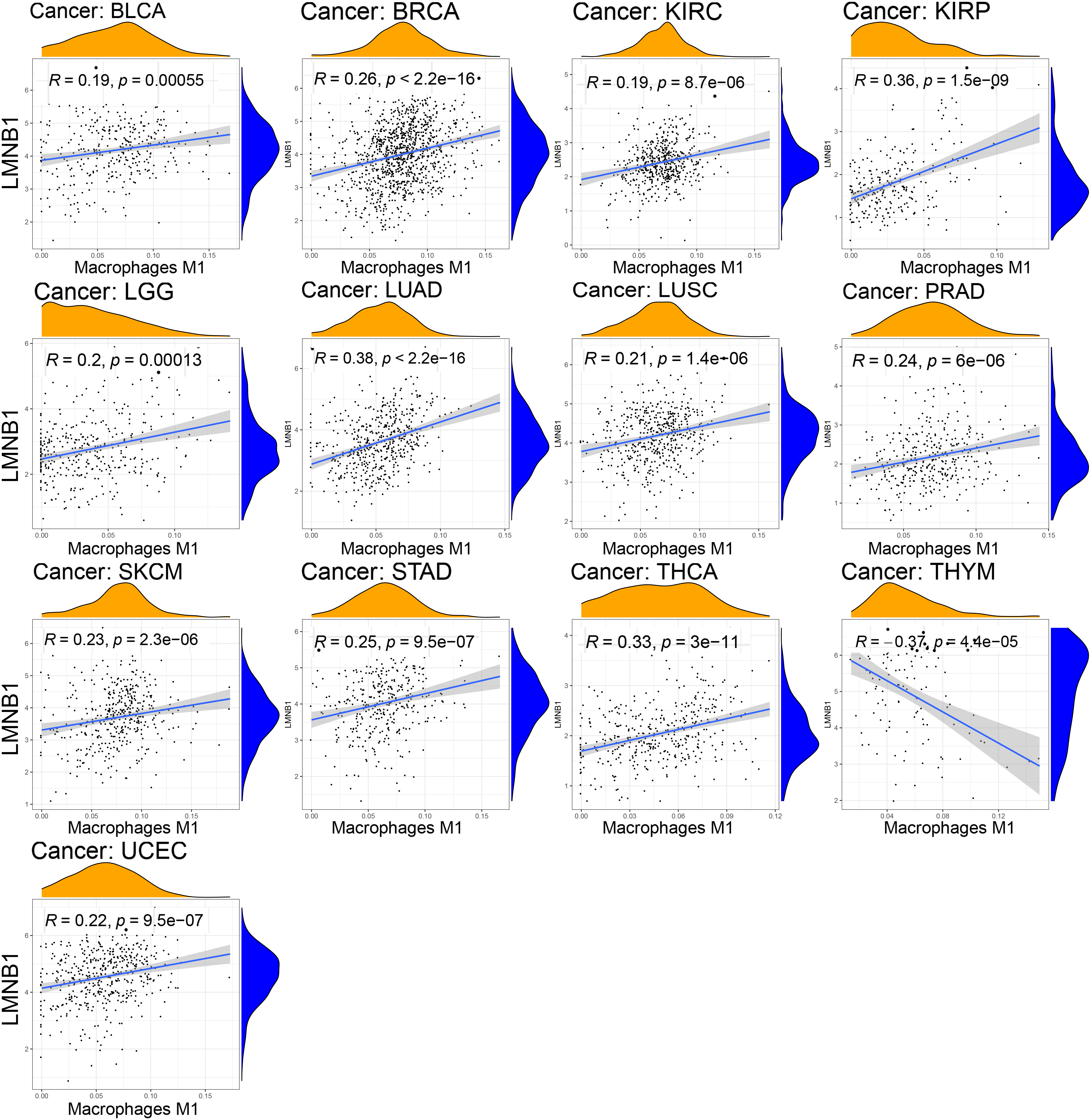
Relationship between the infiltration level of T cells CD4 memory activated and LMNB1 expression. The correlation coefficients and P-values were presented. For scatter plots of each cancer, only scatter plots with 
In GO enrichment analysis of LMNB1, enrichment plots were received for all the 33 cancer types. We counted the enrichment numbers of these pathways based on enrichment plots. Figure 12A showed the top 5 pathways with the highest frequency of occurrence in the enrichment plots. The statistical results suggested that the effect of LMNB1 on tumor pathogenesis might be mainly enriched in “MRNA binding”, “gene silencing”, and “gene silencing by RNA”. In KEGG pathway analysis of LMNB1, enrichment plots were obtained for 10 cancers. We also counted the enrichment numbers of these pathways based on enrichment plots for the 10 cancer types. Figure 12B showed the top 5 pathways with the highest frequency of occurrence in the enrichment plots. The statistical results demonstrated that LMNB1 might influence tumor pathogenesis mainly via “olfactory transduction”, “retinol metabolism”, and “drug metabolism other enzymes”.
Result of enrichment analysis of LMNB1 in 33 cancers. (A) Top 5 pathways with the highest frequency of occurrence in the enrichment plots of GO enrichment analysis. (B) Top 5 pathways with the highest frequency of occurrence in the enrichment plots of KEGG pathway analysis.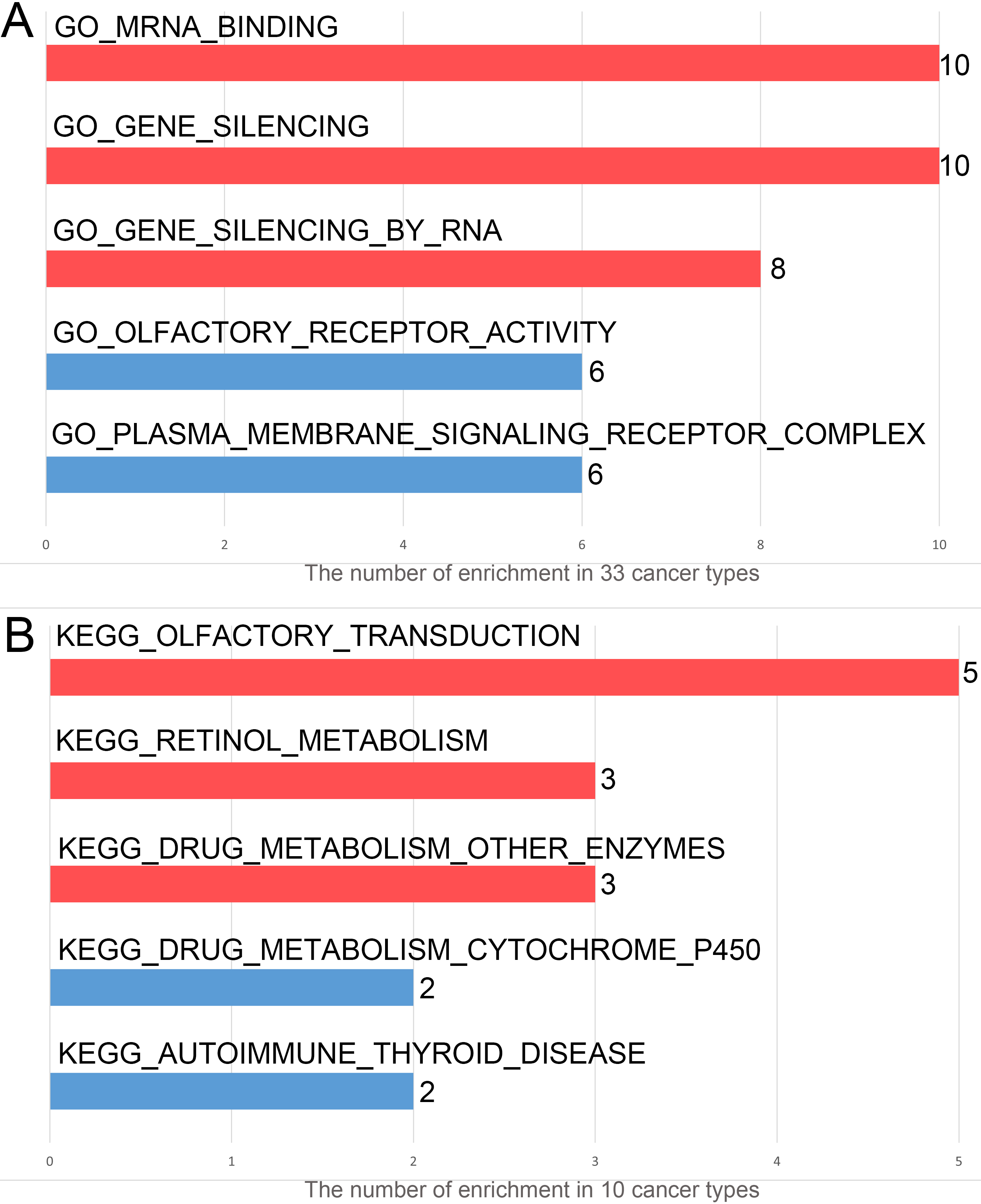
ESCA is a common malignant tumor of human digestive system worldwide and one of the cancers of high incidence [2, 4]. Therefore, it is worth finding a promising predictive biomarker and molecular target for this cancer. In this work, we found the expression and survival prognostic value of LMNB1 in ESCA based on TCGA database. Furthermore, the study of LMNB1 from the perspective of overall tumors has not been reported. Thus, based on the TCGA database, combining with the analyses of gene expression, survival prognosis, TMB and MSI scores, immune infiltration, and gene enrichment, we conducted a comprehensive detection of LMNB1 in human pan-cancer.
In this study, we found that LMNB1 was significantly upregulated in 19 cancer types including ACC, BRCA, COAD, ESCA, GBM, KIRC, LGG, LIHC, LUAD, LUSC, OV, PAAD, PRAD, READ, SKCM, STAD, TGCT, UCEC, and UCS. Further, our study found that the expression of LMNB1 was significantly different between the different pathological stages of 11 cancer types including ACC, BRCA, COAD, ESCA, KICH, KIRC, KIRP, LUSC, SKCM, TGCT, and THCA. These findings manifested that LMNB1 was overexpressed in a variety of tumors and might play a key role in the active development of various tumors. Therefore, it is worth exploring the specific functional role of LMNB1 in cancers further from the perspective of molecular biology experiments. As manifested in several recent molecular biology experiments studies, LMNB1 expression is closely related to nuclear autophagy, the DNA-damage repair, cell death, AKT phosphorylation cell proliferation, cell apoptosis, cell cycle and cell motility in some cancers. For instance, the high expression of LMNB1 mediated by miRNAs promoted the serum starvation-induced nuclear autophagy and accelerated the TAX-induced DNA-damage repair in cervical cancer cells [23]. Likewise, upregulation of LMNB1 also induced dose-dependent cell death and caused the inhibition of colon adenocarcinoma cells migration [24]. On the contrary, knockdown LMNB1 could inhibit the proliferation of LUAD cells by inhibiting AKT phosphorylation [25]. In addition, depletion of LMNB1 inhibited development of LUAD through regulating cell proliferation, cell apoptosis, cell cycle and cell motility [26] These previous studies indicated that LMNB1 expression was tightly associated with tumor development, and the way it affected tumor progression was partly different in different tumors. Therefore, it is of great significance to study the molecular mechanism of LMNB1 in tumor evolution in these 19 tumors with high expression of LMNB1 which may promote LMNB1 to become a promising predictive biomarker. By and large, our study revealed the overexpression of LMNB1 in a variety of tumors ever further, thereby contributing the development of the follow-up studies of pancancer prognostic markers and targeted therapy.
Our current study also first identified the relationship between the expression level of LMNB1 and pan-cancer prognosis. The high expression of LMNB1 was significantly correlated with decreased OS or DFS in 13 cancers, but correlated with well OS in LUSC and THYM. In other words, LMNB1 played a detrimental prognostic role in 13 cancers, while acted a protective prognostic role in LUSC and THYM. Poor prognosis significance of LMNB1 in LIHC, PAAD, and PRAD was consistent with current findings from other studies [27, 28, 29]. In recent studies, upregulation of LMNB1 is associated with poor survival outcomes in patients of KIRC and LUAD [30, 31]. On the contrary, LMNB1 expression was not significant correlated with prognosis of cases with KIRC and LUAD in our findings. These studies used online tool Kaplan–Meier plotter or the “survival” package through R software for survival analysis, while we used the GEPIA2 online analysis database for survival analysis. It needs to be realized that the nuances in the analysis methods and the peculiarities of each data collection and processing methods may exist in different analysis tools. Thereby, these contradictory results may be due to the different tools in survival analysis. Moreover, considering the expression level of LMNB1 in pan cancer, for the 7 cancers including ACC, ESCA, LGG, LIHC, LUSC, PAAD, and PRAD, the LMNB1 expression was not only related to the prognosis of patients, but also significantly different between tumor tissues and the corresponding normal tissues. Hence, in these cancers, it is valuable to further confirm the molecular function of LMNB1 based on molecular cell experiments, which is beyond the scope of our study. However, there is almost still a blank area of research on this issue in these cancers. Consequently future study can focus on the molecular function of LMNB1 in these cancers to develop a novel diagnostic marker. On the whole, our results support the proposition that LMNB1 has strong clinical predictive significance in pan-cancer.
Nevertheless, it was worth noting that there was a different result regarding the expression and prognosis of LMNB1 in LUSC. LMNB1 was dramatically upregulated in LUSC tumor tissues compared to the normal tissues. However, high expression of LMNB1 indicated well OS for LUSC patients. The possible reason was that LMNB1 might play a dual regulatory role in LUSC tumor pathogenesis. To elucidate this phenomenon, future investigation could be explored in specimens to analyze the relationship between expression of LMNB1 and clinical histopathological features such as tumor size, stage, and differentiation in LUSC patients.
Additionally, we found a potential correlation between LMNB1 expression and MSI and TMB for these tumors for the first time. Previous studies have correlated TMB and MSI to patients’ drug responses, especially for drugs that target immune checkpoint inhibitors [32, 33]. In our research results, LMNB1 expression was dramatically correlated with the TMB or MSI for 16 cancer types including ACC, BLCA, BRCA, COAD, ESCA, KICH, KIRC, LGG, LUAD, LUSC, PAAD, PRAD, SARC, STAD, THYM, and UCEC. In fact, both TMB and MSI can be used as predictive factors for the potential efficacy of immunotherapy. For instance, it has been well shown that COAD patients with high MSI demonstrated better checkpoint inhibitor responses and survival in both low and high clinical stages [34]. Similarly, as discovered by Xu et al, LUSC patients with a higher TMB were much more sensitive to immunotherapy and chemotherapy [35]. In our study, both TMB and MSI of 5 cancer types including COAD, LUSC, SARC, STAD, and UCEC were positively related to LMNB1 expression. Even though, further studies are required to confirm whether LMNB1 can act as a good indicator for potential drug responses in these cancers. However, taken together, our findings provide insights into the role of LMNB1 in immunotherapy and drug responses.
In addition, our results confirmed for the first time the relationship between LMNB1 expression and immune infiltration. In 17 cancers of BLCA, BRCA, COAD, HNSC, KIRC, KIRP, LGG, LIHC, LUAD, LUSC, PRAD, SKCM, STAD, THCA, THYM, UCEC, and UVM, LMNB1 expression was significantly correlated with the infiltration level of Macrophages M1 or T cells CD4 memory activated. Previous studies have shown that immune cells played an indispensable role as a double-edged sword in tumors to promote or inhibit tumor progression [36, 37]. As fundamental members of the immune cells, Macrophages M1 is highly specialized in programmed cell removal and tends to recognize and capture tumor cells, while T cells CD4 memory activated inhibits the growth of tumor cells by promoting the proliferation of T cells CD8 [38, 39]. Therefore, the results in this study suggested that LMNB1 might indirectly lead to tumorigenesis or inhibit the growth, invasion, and metastasis of tumor cells by regulating the infiltration level of different immune cells in tumor immunity for these cancers, especially for BLCA, BRCA, KIRC, KIRP, LUAD, PRAD, STAD, THCA, UCEC. These preliminary findings constitute substantial advance toward the identification of a nonnegligible role for LMNB1 in tumor infiltration.
Further, we counted the results of the enrichment analysis of LMNB1 in pan-cancer. Our gene enrichment research results indicated that LMNB1 might not only participate in the pathways including “olfactory transduction”, “retinol metabolism”, and “drug metabolism other enzymes”, but also participate in the cellular biological processes such as “gene silencing”, “MRNA binding”, and “gene silencing by RNA”. As demonstrated by several well reports about the relationship between these pathways and cancers, 9-cis-retinol in combination with cRDH inhibited breast cancer cell proliferation by production of retinol metabolites [40]. And gene silencing was correlated to promoter hypermethylation which had an essential role on tumor progression [41]. In addition, some mRNA binding protein could promote cell proliferation and invasion [42, 43, 44]. These studies indicated that retinol metabolism, gene silencing, and mRNA binding protein promoting cell proliferation and invasion might be key events in the development of multiple cancer types. However, further verification is needed to confirm whether LMNB1 can affect tumor progression through these pathways and cellular biological processes. Future study may focus on the relationship between LMNB1 expression and the above pathways and cellular biological processes. In a word, these novel findings indicate a new orientation for studying the potential roles of LMNB1 in the neoplastic process.
Although our study has provided useful evidence of involvement of LMNB1 in tumorigenesis and regulation of the immune environment in tumor cells, it does comprise some limitations. First, it is a pure bioinformatics analysis completely dependent on information available in open-access databases without confirmation through experimentation. Second, as a B-type lamin, LMNB1 needs more further studies to determine its biomarker utility in many cancers, due to its variable expression patterns between and within cancer subtypes [45].
In summary, our WGCNA-based pan-cancer analysis of LMNB1 systematically demonstrated that LMNB1 was not only differentially expressed in a variety of tumors, but also significantly correlated with patients’ prognosis, TMB, MSI and immune infiltration in multiple cancers. In addition, its enrichment pathways were also related to tumor progression. Therefore, it revealed its up-regulation and potential clinical prognostic value in human pan-cancer, thereby providing the first-hand data to understand the role of LMNB1 in various cancers from the perspective of tumor specimen. Meanwhile, these findings may provide preliminary insights for further exploration of its mechanism in pan-cancer and confirmation of its biomarker utility in pan-cancer from the perspective of molecular biology experiments and thereby will be conducive to promote its development into a valuable novel target for cancers.
Author contributions
Youwei Hua: conception, interpretation or analysis of data, preparation of the manuscript, revision for important intellectual content.
Zhihui He: data validation and manuscript revision.
Xu Zhang: conception, revision for important intellectual content, supervision.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-203247.
sj-docx-1-cbm-10.3233_CBM-203247.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-203247.docx
Footnotes
Acknowledgments
This study was supported by the Natural Science Foundation of China (No. 11701471,