
Abstract
PURPOSE:
Aberrant DNA methylation plays a crucial role in oral carcinogenesis. Our previous study demonstrated hypermethylation of DAPK1, LRPPRC, RAB6C, and ZNF471 promoters in patients with tongue squamous cell carcinoma compared with normal samples. Methylation profiling using salivary DNA is considered a non-invasive alternative to tissue samples. Hence, the present study tested the DNA methylation status of these four promoters as indicators of oral cancer progression.
METHODS:
We performed the bisulfite-based targeted next-generation sequencing of four candidate genes in saliva and tissue DNA from normal, premalignant, and squamous cell carcinoma subjects. The clinicopathological association, diagnostic, and prognostic utility of aberrant DNA methylation were evaluated using the TCGA-HNSCC dataset. Using the Xgboost algorithm and logistic regression, CpG sites were prioritized, and Receiver Operating Characteristic was generated. By Log-rank test and Kaplan-Meier (KM) curves, an association between methylation and overall survival (OS), disease-free interval (DFI), and progression-free interval (PFI) were computed.
RESULTS:
We identified all four genes as significantly hypermethylated in premalignant and malignant samples compared with normal samples. The methylation levels were comparable between saliva and tissue samples with an r-value of 0.6297 to 0.8023 and 0.7823 to 0.9419 between premalignant tissue vs. saliva and OC vs. saliva, respectively. We identified an inverse correlation between DAPK1, LRPPRC, RAB6C, and ZNF471 promoter methylation with their expression. A classifier of 8 differentially methylated CpG sites belonging to DAPK1, RAB6C, and ZNF471 promoters was constructed, showing an AUC of 0.984 to differentiate tumors from normal samples. The differential methylation status of DAPK1, LRPPRC, and ZNF71 promoters was prognostically important. Abnormal expression of all four genes was associated with immune infiltration.
CONCLUSIONS:
Thus, methylation analysis of these candidate CpG sites from saliva can be helpful as a non-invasive tool for the clinical management of OC.
Keywords
Introduction
Oral cancer (OC) is a major public health problem that accounts for 1.8% of all cancer deaths [1]. Lip and oral cancer accounted for 377,713 new cases and 177,757 deaths in 2020 [1]. The use of tobacco in smoking, chewing pan, and alcohol consumption are the risk factors for OC [2]. Epidemiological studies causally associated human papillomavirus (HPV) infection with OC [3]. Despite the availability of advanced early detection tools, OC is detected late, contributing to the high treatment cost, adverse clinical outcomes, and high mortality rate [4]. Identifying the early molecular changes may improve the clinical management of OC by providing markers for early diagnosis, prognosis, and therapy response.
Aberrant DNA methylation is linked to features that underlie many phenotypic changes in OC pathogenesis that could serve as potential markers for the detection and prognosis [5, 6, 7]. Quantifying methylation differences at specific sites could delineate a clinically significant threshold to distinguish the disease states in an evolving tumor and establish signature epigenetic marks for early detection [8]. Methylation profiling using DNA isolated from buccal swabs, oral rinse, or saliva suggested the usefulness of these samples as a non-invasive alternative to tissue samples [9, 10]. Further, salivary methylation profiling was demonstrated as a non-invasive and reliable alternative for tissue biopsy samples for the clinical management of OC [10].
Previously we have reported that the CpG site belonging to Death-Associated Protein Kinase 1 (DAPK1), Leucine-Rich Pentatricopeptide Repeat Containing (LRPPRC), Ras-related Protein Rab-6C (RAB6C), and Zinc Finger Protein 471 (ZNF471) promoters are hypermethylated in a cohort of tongue squamous cell carcinoma patients when compared with normal samples [8]. Herein, the promoter DNA methylation status of four candidate genes was evaluated using saliva and tissues collected from the normal, premalignant lesion, and OC patients. The findings were confirmed by analyzing the methylation levels of candidate genes from The Cancer Genome Atlas head and neck squamous cell carcinoma (TCGA-HNSCC) dataset [11]. We next tested these candidate genes’ diagnostic and prognostic utility in OC. We observed that the methylation status of four genes in salivary DNA was comparable to those of the premalignant and malignant tumor tissue specimens. We identified that aberrant methylation of four genes was (i) linked with high sensitivity and specificity to differentiate tumors from normal samples and (ii) potential prognosticator for OC. Our study showed an association between the methylation status of DAPK1, LRPPRC, RAB6C, and ZNF471 gene promoter as surrogate non-invasive markers for the early diagnosis of malignant transformation of the potentially malignant lesion to OC.
Materials and methods
Sample collection
The Institution Ethics Committee approved the study. The samples were collected only after written consent from the participants. Biopsy specimens from normal, premalignant, and squamous cell carcinoma subjects (
DNA extraction
QIAamp
Bisulfite conversion and PCR
Bisulfite conversion of DNA (0.5 to 1
Next-generation sequencing
We used NGS to identify the differently methylated CpG sites within a given amplicon, as published previously [12, 13]. Ion Xpress™Plus Fragment Library Kit was used for library preparation from purified PCR products. PCR products of four genes/samples were pooled, fragmented using Ion Shear™Plus Reagents, purified using Agencourt AMPure XP beads (Beckman Coulter, USA), and ligated to an Ion P1 adapter. The individual samples were barcoded with the Ion Xpress barcode. Following purification by Agencourt AMPure XP beads, 330bp target library (200base read length) was selected with 2% agarose gel (E-Gel SizeSelect™), amplified using Ion Xpress™Plus Fragment Library Kit. The quality and quantity of the amplified library were analyzed using the High Sensitivity DNA kit and Qubit
Analysis of NGS data
The NGS data was processed, and differentially methylated CpG sites were identified, as published earlier [12, 13]. The FastQC files were used for analyzing the quality of raw reads, followed by their alignment with reference sequence using NextGenMap [14]. BiQAnalyzer HT software (
In silico analysis
The methylation status of four candidate genes in the TCGA-HNSCC dataset was analyzed using DNMIVD (
Diagnostic model construction
As published earlier, a diagnostic model was constructed using the DNMIVD tool [16]. A cluster map, Receiver Operating Characteristic (ROC) curve, and Area Under Curve (AUC) was constructed to differentiate tumor from normal samples. Feature importance scores for individual CpGs were computed using the xgboost algorithm [16]. The logistic regression model was implemented to generate the diagnostic model and predict AUC via ROC curve construction.
Prognostic model construction
As published earlier, the prognostic model was constructed using the DNMIVD tool [16]. The samples were partitioned into low and high methylation groups using default parameters. The Log-rank test and Kaplan-Meier (KM) curves were generated to predict the overall survival (OS), disease-free interval (DFI), and progression-free interval (PFI).
Immune infiltration analysis
We have used the TIMER (
Statistical analysis
The average methylation was calculated by the sum of the methylation levels of all the CpG sites analyzed divided by the total number of CpG sites multiplied by 100. GraphPad Prism (version 7) was used for statistical analyses. The clinical significance of hypermethylation was evaluated by Fisher’s two-tailed tests and paired
Heatmap representing methylation levels of (a) DAPK1, (b) LRPPRC, (c) RAB6C, and (d) ZNF471 by bisulfite-based targeted next-generation sequencing in normal, premalignant, OSCC (
Demographic characteristics of the study population
The median age of premalignant, OC and normal subjects was 56
DNA Methylation status of candidate genes in normal, OPMD, and OSCC cases
The level of DNA methylation of the four gene promoters in normal, premalignant, and OC samples were tested by NGS on DNA extracted from saliva and tissue samples. The heat map representing the methylation levels of DAPK1, LRPPRC, RAB6C, and ZNF471 is shown in Fig. 1. The methylation frequency of all the four-gene promoters was significantly higher in premalignant and OC compared with controls samples (Supplementary Table 3,
The promoter methylation analysis of four candidate genes showed significant hypermethylation of DAPK1, LRPPRC, and ZNF471 in premalignant and OSCC samples compared to healthy normal control samples (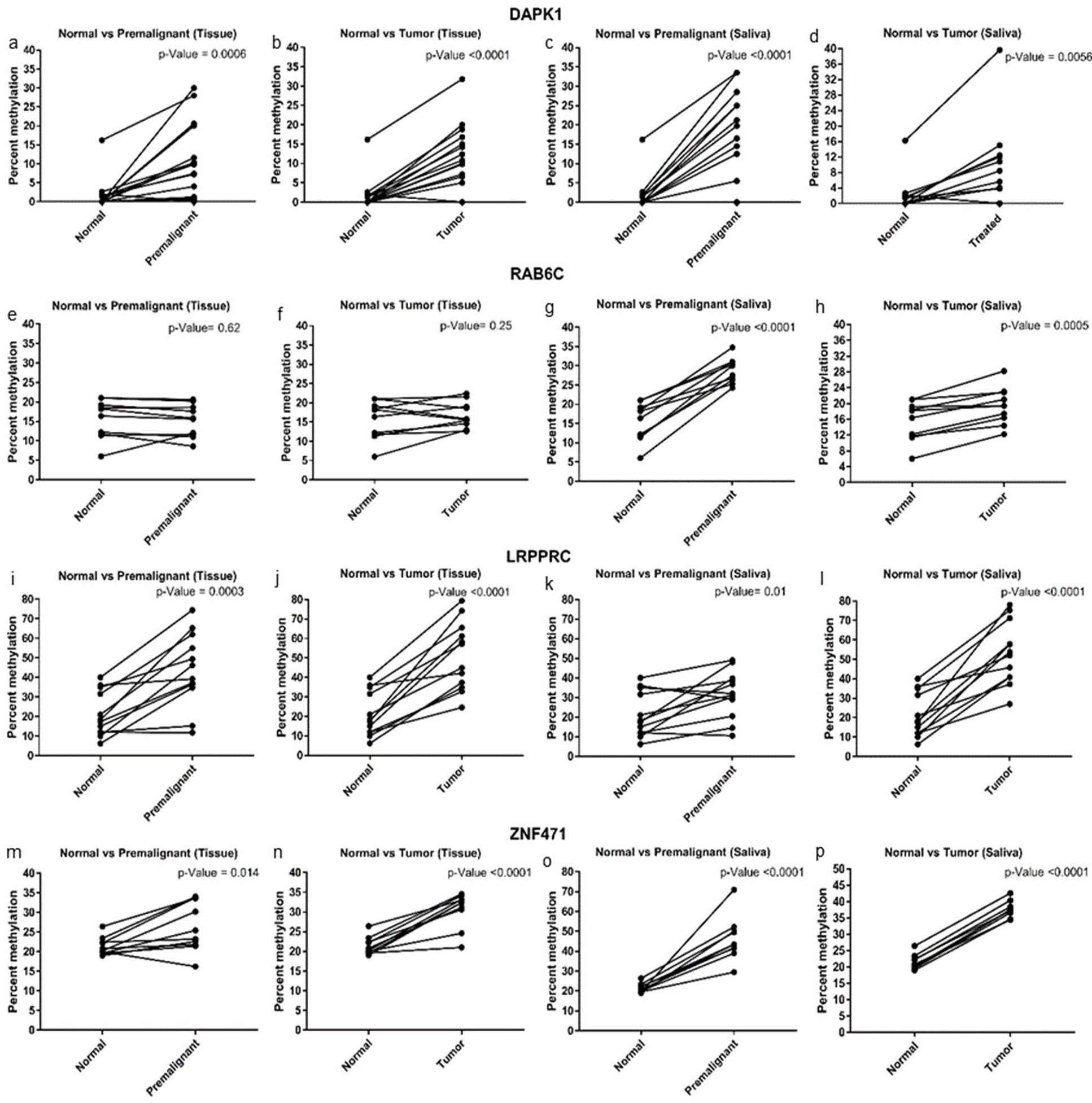
The average methylation level for any individual gene studied was higher in OPMD and OSCC (tissue and saliva) compared to normal samples (Supplementary Table 3). Figure 2 represents the average promoter methylation of the four candidate genes in tissue and saliva collected from normal, premalignant, and tumor samples. DAPK1, LRPPRC, and ZNF471 promoters showed significant hypermethylation in premalignant and OSCC samples compared to healthy normal controls (Fig. 2). RAB6C promoters showed hypermethylation in premalignant, and OSCC saliva but not in tissue samples (Fig. 2). Individual CpG site analysis showed significantly higher promoter methylation for the gene promoters analyzed in OPMD and OC samples than in normal samples (Fig. 3).
The individual CpG sites of four candidate genes showed significant differential methylation as analyzed by two-way ANOVA analysis (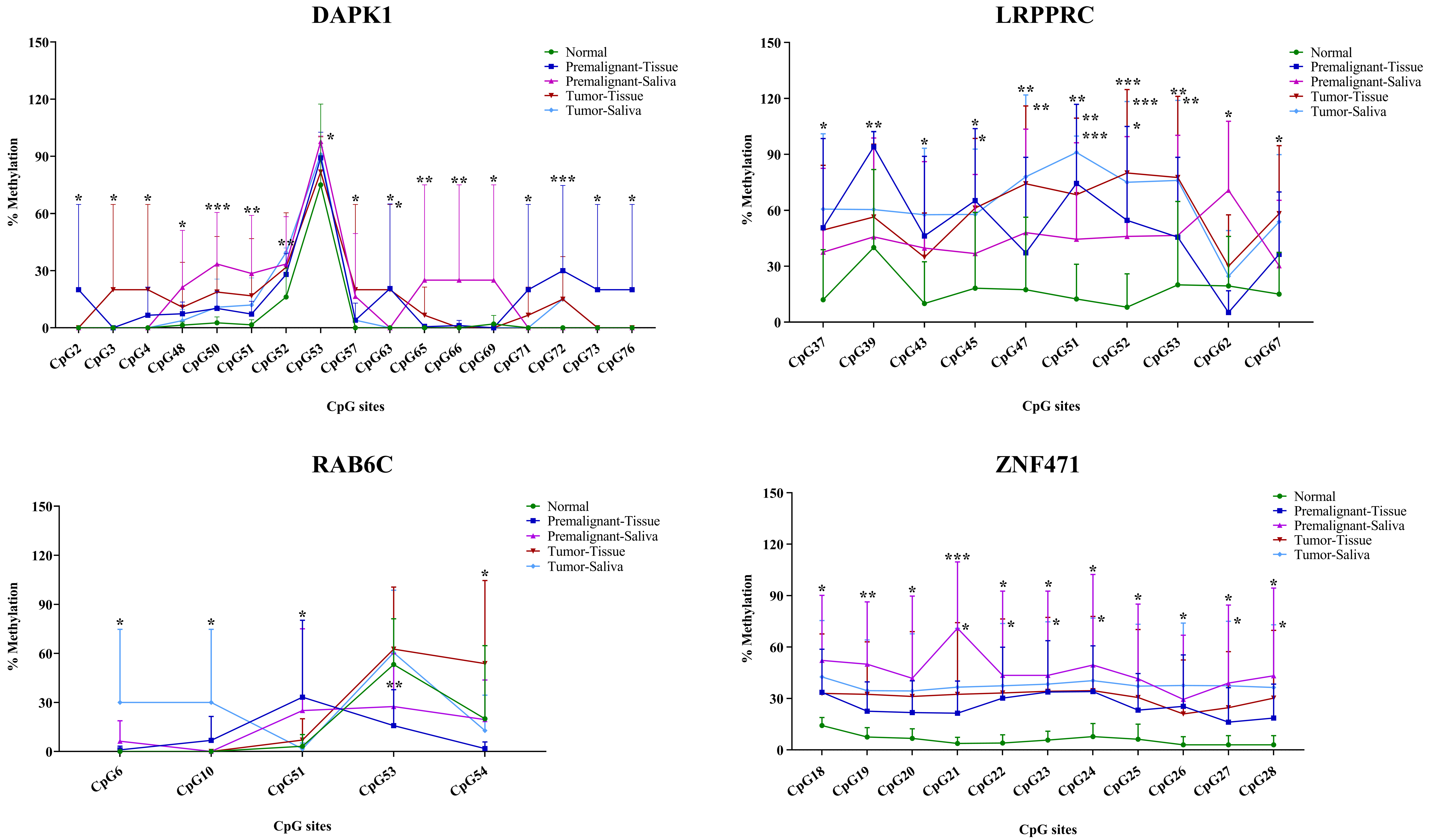
Within a given individual, the site-specific methylation levels were positively correlated in both saliva and tissue samples as analyzed by Pearson’s correlation analysis(Supplementary Table 4,
DAPK1, LRPPRC, RAB6C, and ZNF471 promoters are abnormally methylated in TCGA- HNSC dataset
Since our sample number was small, we analyzed the methylation status of four gene promoters in the oral cancer samples (normal
The promoter methylation status of four candidate genes in the oral cancer samples in the TCGA-HNSCC dataset (a) The heat map showing the methylation level of 34 normal and 318 oral cancer samples. The normal and tumor samples showed distinct methylation patterns for all the four tested genes (b) Dot plot representing the methylation status of four genes in 34 normal and 318 oral cancer samples. DAPK1, LRPPRC, RAB6C, and ZNF471 were significantly hypermethylated in oral cancer samples compared with normal samples.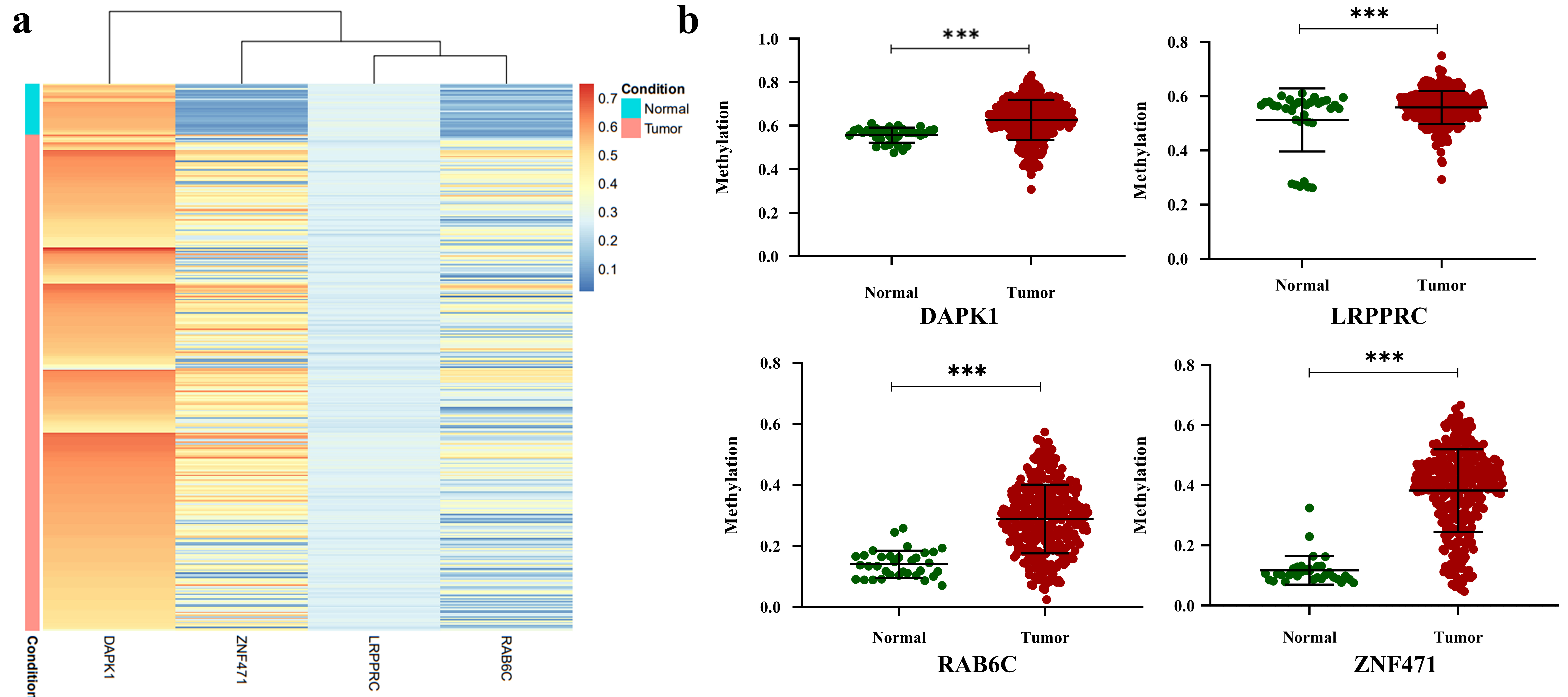
We next tested the association between promoter methylation of four test genes with clinicopathological characteristics such as stage, grade, race, gender, smoking, metastasis, and p53 status in the TCGA-HNSC dataset(Supplementary Table 5). The methylation levels of DAPK1, LRPRC, and ZNF471 were significantly different between normal vs. stage 1 and stage 4 cancers. However, the RAB6C promoter was significantly differentially methylated between normal vs. stage 2 and stage 4 cancers. All four-gene promoters’ differential methylation status was significantly different in Caucasians and African Americans compared with normal. More interestingly, among the cancer patients, DAPK1 methylation was significantly different between male and female patients. All four genes were significantly differentially methylated between normal vs. grade 1 to grade 4 patients. More specifically, DAPK1 showed methylation differences for Grade 1vs. Grade 3, Grade2 vs. Grade3, Grade3 vs. Grade4- tumors. RAB6C methylation could differentiate Grade1 vs. Grade2, Grade1 vs. Grade4, and Grade2 vs. Grade4 tumors. ZNF471 methylation was significantly different between Grade1 vs. Grade 2 tumors.
Methylation of LRPPRC and RAB6C was significantly different between Non-Smoker vs. Smoker. DAPK1 methylation was significantly different between Non-Smoker vs. Reformed Smoker1. ZNF471 methylation was significantly different between Smoker and Reformed Smoker1. LRPPRC and RAB6C showed significant differential methylation in Smoker vs. Reformed Smoker 2. For nodal metastasis, the methylation level was significantly different from LRPPRC and ZNF471 for N0 vs. N2. Furthermore, the methylation status of DAPK1 and LRPPRC were significantly different between TP53 Mutant vs. TP53 Non-Mutant OC.
The methylation of 8 CpG sites can distinguish tumor from normal samples (a) Clustered map of 8 differentially methylated CpG sites DAPK1, RAB6C, and ZNF471 promoter. (b) The Xgboost and logistic regression analysis shortlisted 8 out of 33 CpG sites with diagnostic importance. (c) The methylation status of 8 CpG sites showed a ROC of 0.984.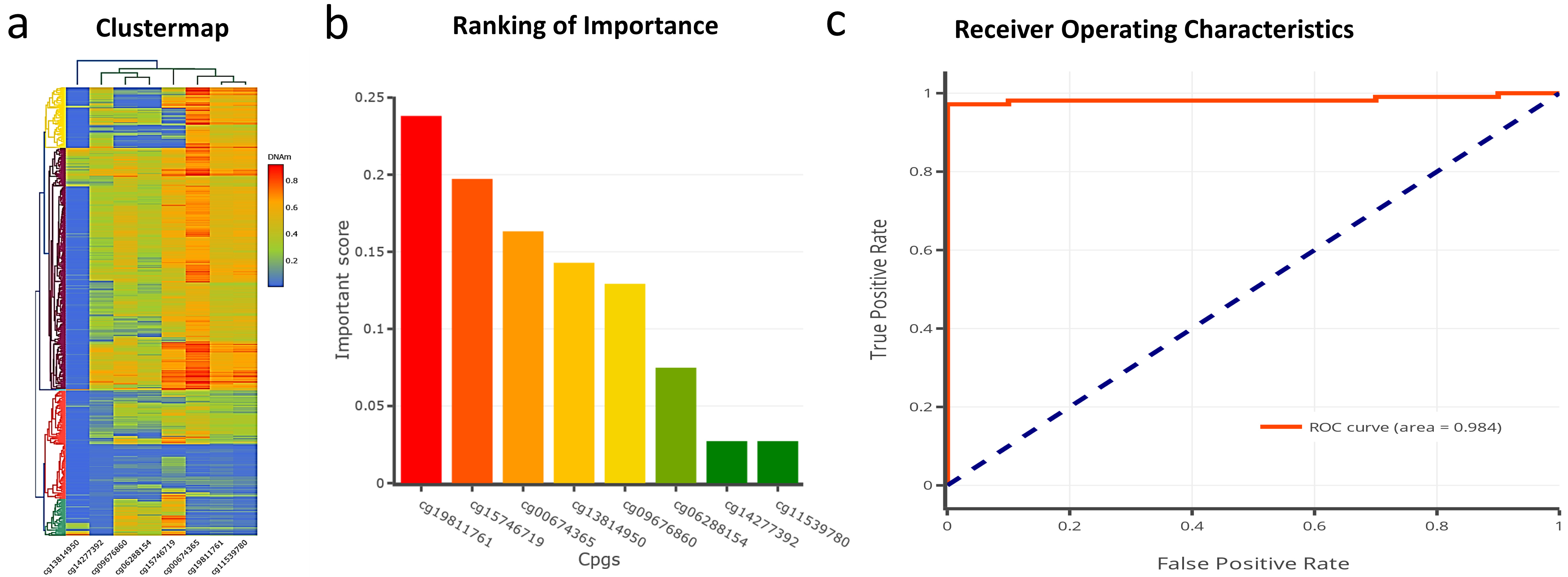
We next investigated the diagnostic significance of the promoter methylation status of 4 genes for diagnosing HNSCC using the TCGA-HNSC dataset. First, the clinically relevant differentially methylated CpG sites were computed using the xgboost algorithm to build a diagnostic model (Fig. 5a and b). Figure 5a represents the cluster map of 8 hypermethylated CpG sites. Figure 5b represents the key CpG sites with the potential to differentiate tumors from normal samples identified via the xgboost algorithm. The xgboost algorithm and logistic regression analysis of 8 hypermethylated CpG sites belonged to DAPK1, RAB6C, and ZNF471 promoter differentiated tumors from normal samples with an AUC of 0.984 (Fig. 5c).
Differential methylation of DAPK1, LRPPRC, and ZNF71 are prognostically important
Using the Log-rank test, we analyzed the prognostic significance of differential methylation of four candidate gene promoters in HNSCC using the TCGA-HNSCC dataset(Supplementary Fig. 2). Among the four genes tested, the hypermethylation of ZNF471 was associated with poor OS. The methylation status of DAPK1, LRPPRC, RAB6C, and ZNF471 did not show any significant association with PFI. However, the methylation status of DAPK1 and LRPPRC genes was associated with DFI.
DAPK1, LRPPRC, RAB6C, and ZNF471 expression correlated with immune infiltration
The TIMER tool was used for immune infiltration. In HNSC, all four gene expressions positively and negatively correlated with CD4
Discussion
OC is among India’s critical public health problems and many other countries [1]. Cancer detection at an advanced stage and lack of reliable prognostic markers contribute to OC’s high incidence and mortality [2, 4, 20]. Changes in the DNA methylation status have been shown to occur at an early stage of tumor development, highlighting the importance of epigenetic modification in OC’s initiation, development, and progression [5]. Hence, assessing the extent of methylation in the CpG-rich regions and functional characterization of downstream targets may unravel the molecular events facilitating carcinogenesis. Besides, such changes may be helpful in diagnosis, prognosis, and improved management of OC.
This study systematically analyzed the relationship of four gene promoter methylation in normal, premalignant, and SCC using saliva and tissue samples, followed by an independent confirmation of our findings using TCGA-HNSCC datasets. We demonstrated that all the genes promoter tested were hypermethylated in our samples. The presence of hypermethylated CpG sites within ZNF47, RAB6C, LRPPRC, and DAPK1 promoters in the premalignant condition suggests that the methylation profiling of these sites could be helpful in an early diagnosis of OC. ROC analysis demonstrated that the methylation of 8 CpG belonging to ZNF471, RAB6C, and DAPK1 could help distinguish tumors from normal samples. The differential promoter methylation of four genes also correlated with immune infiltrations. Collectively, our data demonstrated that DAPK1, LRPPRC, RAB6C, and ZNF471 are differentially methylated between normal, premalignant, and tumor samples and that abnormal methylation may be helpful as a diagnostic and prognostic indicator in OC.
The present study is the first to describe the hypermethylation of ZNF471, RAB6C, and LRPPRC in saliva. A high positive correlation of DNA methylation profile between saliva and tissue implies the feasibility of saliva as a reliable alternative to oral tissues for methylation analysis in a clinical setting. Our study supports the previous studies supporting saliva as an alternative to tissue for methylation profiling in OC [10, 21, 22]. Hansen and co-workers showed that the response rate of participants to provide saliva was significantly higher (72%) than blood samples (31%) [21]. This is particularly beneficial to patients in clinical settings where salivary methylation profiling could be an adjunct diagnostic tool for screening, monitoring, and patient follow-up.
DAPK1, located at 9q21.33, encodes for a 160kD tumor suppressor protein that is downregulated in OC via hypermethylation [23]. It is a calcium-calmodulin-dependent serine-threonine kinase linked with pathways such as cell survival, type 1 apoptosis, and type II autophagy [24]. Downregulation of DAPK1 induces cell proliferation and growth in multiple cancer, suggesting it is a tumor growth regulator gene [25, 26, 27]. Inactivation of DAPK1 by hypermethylation is reported in metastatic progression in multiple cancer types [28, 29]. Tumor-specific DAPK1 hypermethylation is reported using DNA isolated from saliva in HNSCC [30]. A study by Dmitry and co-workers showed that salivary methylation profiling for RASSF1A, DAPK1 and p16 genes showed a tumor-specific accuracy of 81% in HNSCC [30]. A comprehensive meta-analysis showed that the aberrant methylation of DAPK1 promotes in HNSCC [23]. Our study showed that differential methylation of DAPK1 can help differentiate grading the tumor samples. Interestingly, the promoter methylation of DAPK1 was also significantly different between p53 wild type vs. p53 mutant tumor samples.
ZNF471, located at 19q13.43, encodes for a 73kD protein proposed to possess DNA-binding transcription factor activity and is frequently hypermethylated and silenced in a few cancers [8, 12, 31, 32, 33]. We have previously reported the hypermethylation of ZNF471 for the first time in SCCT [8]. The TCGA-HNSCC data set analysis demonstrated a strong negative correlation between promoter methylation and ZNF471 expression, suggesting that ZNF471 may be a methylation-regulated gene in OC. Besides, hypermethylation of ZNF471 also correlated with poor OS. ZNF471 methylation profiling could help differentiate smoker vs. reformed smoker 1 and N0 vs. N2 nodal metastasis. Hence, more detailed functional and mechanistic investigations are required to understand the contribution of ZNF471 in OC.
LRPPRC located at 2p21 encodes for a 157.9kD leucine-rich protein having multiple pentatricopeptide repeats is suggested to take part in transcriptional regulation of nuclear and mitochondrial encoded genes [34]. It is predicted to contain a mitochondrial targeting sequence at its N-terminal and is predominantly localized to mitochondria [35]. Increased LRPPRC expression is reported in lung, gastric, and prostate cancer and correlated with disease progression [35, 36, 37, 38]. Inhibition of LRPPRC has been shown to inhibit growth and invasion with concomitant induction of apoptosis [38]. LRPPRC is an overexpressed gene in gastric cancer, and its forced downregulation reversed therapy resistance [33]. LRPPRC is overexpressed in retinoblastoma and promotes migration, invasion, and glycolysis via autophagy suppression 3̧9. LRPPRC promoted cisplatin resistance in lung cancer [36]. Our study reported the hypermethylation of LRPPRC in saliva and OC samples. LRPPRC hypermethylation did not influence OS and PFI. However, LRPPRC hypermethylation prolonged the DFI in TCGA-HNSCC dataset. More detailed mRNA or protein level expression studies are needed to understand the role of LRPPRC hypermethylation in prolonging the survival of HNSCC patients. Towards this, its contribution to OSCC requires further studies. A comprehensive mechanistic studies are needed to understand the role of LRPPRC in OC.
RAB6C is located in 2q21.1 and encodes for a 28.35kD protein detected as hypermethylated in our samples and the TCGA-HNSCC dataset. In breast cancer, hypermethylation and downregulation of RAB6C are linked with the induction of multidrug resistance [40]. RAB6C is an inhibitor of migration, invasion, and metastasis in breast cancer [41]. The ER
With the progress in immunotherapy and success with immune checkpoint inhibitors in cancer, researchers are increasingly focusing on the role of abnormal expression of the genes in immune infiltration and cancer progression [43]. We used TIMER online database to find the possible association of the four candidate genes with immune infiltration. Our analysis showed that altered expression of all four genes in HNSCC was correlated with CD4
Our study had several limitations. First, the methylation analysis was performed on only a small amount of saliva. Second, we did not perform the expression analysis of the four-candidate gene. Third, detailed functional and mechanistic studies are needed to understand the biological effect of these candidate genes. Finally, many clinical correlation analyses used TCGA-HNSCC data sets via freely available online tools. However, it is worth noting that the associations between aberrant methylation of DAPK1, LRPPRC, RAB6C, and ZNF471 with the development and progression of OC and salivary DNA methylation may be helpful for early diagnosis of OC.
Conclusion
The utility of saliva as an alternative tissue source for molecular studies and biomarker discovery is gaining attention. In our study of OPMD and OC subjects, the salivary DNA methylation pattern of the candidate gene promoters was comparable to tumor tissue specimens, thus demonstrating the potential to be used as a surrogate non-invasive marker for the early diagnosis of OC. Our study can potentially establish DAPK1, LRPPRC, RAB6C, and ZNF471 gene promoter methylation analysis as potential biosignatures in a large OSCC clinical cohort to fully establish its diagnostic utility. Although the sample size is small, our prospective study uses a saliva sample, i.e., a non-invasive technique to explore the methylation signatures. Future studies must validate our findings in a larger cohort of patients. Detailed studies are also required to investigate the biological function and cell signaling pathways regulated by these genes in the pathophysiology of OC.
Statements and declarations
The authors declare no conflict of interest. All the authors have read and approved the final draft of the manuscript.
Author contributions
Conception: SPK and AK.
Interpretation Or Analysis Of Data: SC, VKV, SG, RR, and SM.
Preparation Of The Manuscript: SPK, RR, and AK.
Revision For Important Intellectual Content: RR, SPK, and AK.
Supervision: SC, SPK, and AK.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-220028.
sj-pdf-1-cbm-10.3233_CBM-220028.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-220028.pdf
Footnotes
Acknowledgments
This study received funding from the Royal College of Surgeons of Edinburgh under the Small Research Pump Priming Grants scheme (SRG/16/099) awarded to Adarsh Kudva. We acknowledge Dr. TMA Pai Structured Ph.D. fellowship, Manipal Academy of Higher Education (MAHE), for fellowships. All the authors thank the Technology Information Forecasting and Assessment Council (TIFAC)-Core in Pharmacogenomics at MAHE, Manipal, Fund for Improvement of S&T Infrastructure (FIST), and Karnataka Fund for Infrastructure Strengthening in Science and Technology (K-FIST), Government of Karnataka.