
Abstract
BACKGROUND:
With the rapid development of genomics and molecular biology, not only have biochemical indicators been used as tumour markers, but many new molecular markers have emerged. Epigenetic abnormalities are a new type of molecular marker, and DNA methylation is an important part of epigenetics.
OBJECTIVE:
This study used weighted gene coexpression network analysis (WGCNA) to analyse key methylation-driven genes in breast cancer.
METHODS:
The RNA-seq transcriptome data, DNA methylation data, and clinical information data of breast cancer patients were downloaded from The Cancer Genome Atlas (TCGA) database, and the MethylMix R package was used to screen methylation-driven genes in breast cancer. The ClusterProfiler package and enrichplot package in R software were used to further analyse the function and signalling pathway of methylation-driven genes. Through univariate and multivariate Cox regression analyses, methylation-driver genes related to prognostic were obtained, a prognostic model was constructed and prognostic characteristics were analysed.
RESULTS:
The 17 methylation-driven genes related to prognosis were obtained by the WGCNA method in breast cancer, and the prognostic significance of these methylation-driven genes was determined by transcriptome and methylation combined survival analysis. Analysis of functions and signalling pathways showed that these genes were mainly enriched in biological processes and signalling pathway. Through univariate and multivariate Cox regression analyses, a prognostic model of 5 methylation-driven genes was constructed.
CONCLUSIONS:
The AUC of the receiver operating characteristic (ROC) curve of this model was 0.784, showing that the model had a good prediction effect. Based on WGCNA screening, it was found that only CDO1 was the key methylation-driven gene for prognosis in breast cancer, indicating that CDO1 may be an important indicator of the prognosis of breast cancer patients.
Abbreviations
The research route of this article.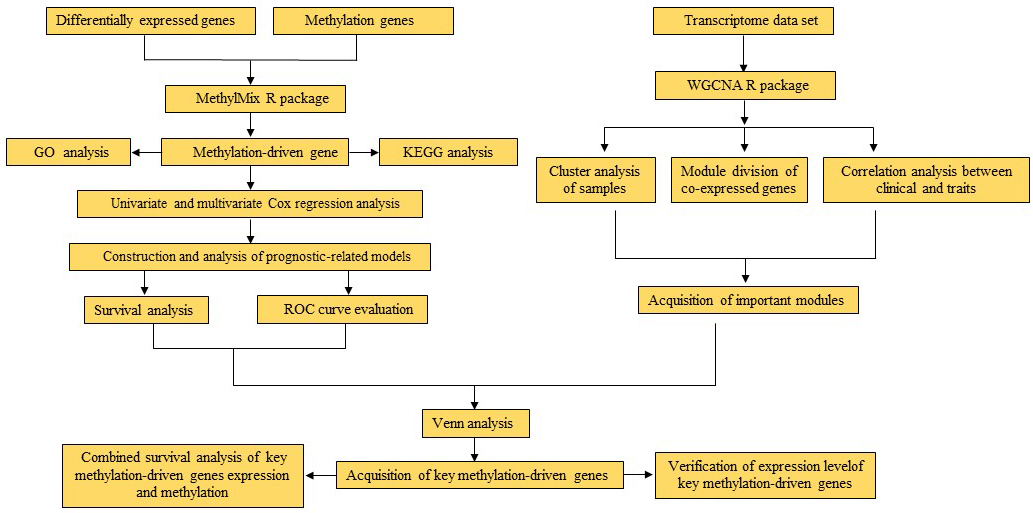
The latest global cancer statistics report shows that there are 2.3 million new cases of breast cancer in the world in 2020, which has replaced lung cancer as the largest cancer in the world, accounting for 11.7% of the incidence of malignant tumors [1]. From a global perspective, the incidence of breast cancer ranks first among female cancers, and approximately 24.5% of new female cancer cases are breast cancer [1]. Currently, tumour screening, diagnosis and treatment methods have been greatly improved. Early breast cancer patients can be effectively cured, but no ideal treatment methods have been established for patients with postoperative recurrence, distant metastasis or advanced breast cancer. Therefore, it is necessary to find an effective method to prevent, diagnose and treat breast cancer to reduce the incidence and mortality of breast cancer. With the rapid development of genomics and molecular biology, not only have biochemical indicators been used as tumour markers, but many new molecular markers have emerged. Epigenetic abnormalities are a new type of molecular marker, and DNA methylation is an important part of epigenetics. The abnormal DNA methylation of tumour cells mainly include hypermethylation and hypomethylation. The overall DNA in tumour cells is in a state of hypomethylation, and part of the promoter region is in a state of hypermethylation. Hypermethylation in the promoter region of tumour suppressor genes will downregulate or hinder the transcription level, and ultimately induce tumorigenesis [2, 3, 4, 5]. Abnormal DNA methylation is a new type of tumour marker that plays an important role in the diagnosis, treatment and prognosis of clinical tumours, and an increasing number of biomarker kits based on methylation have been developed [6, 7]. Moreover, it has been shown that abnormally methylated genes can serve as potential cancer driver [8]. Therefore, it is of great value to study the methylation-driven genes of breast cancer to achieve early diagnosis, identify new therapeutic targets, and improving the survival rate and prognosis of breast cancer patients
Weighted gene co-expression network analysis (WGCNA) is a typical system biology algorithm for constructing gene co-expression network, which can analyze complex and large-scale high-throughput expression microarray data and show the correlation between genes in different samples [9]. At present, WGCNA has been widely used in the international biomedical field, such as the successful identification of cancer biomarkers [10]. Generally, the genes at the center of the regulatory network are called core genes. These genes are usually key regulators such as transcription factors, and their roles are worthy of our priority for in-depth analysis and mining [11].
In this paper, the transcriptome data and DNA methylation data of breast cancer in the TCGA database were combined to screen methylation-driven genes, and a prognostic model was constructed. Then, the key prognostic methylation-driven genes in breast cancer were screened based on the comprehensive method of WGCNA [10], the research route is shown in Fig. 1. Our research will provide new insights into the molecular mechanism of breast cancer and provide new directions for the treatment of breast cancer.
Materials and methods
Data acquisition
The RNA-seq transcriptome data, DNA methylation data, and clinical information of breast cancer were obtained from the TCGA database (
Identification of methylation-driven gene
Sorting out the transcriptome data and methylation data, the differentially expressed genes (DEGs) between tumour and normal samples were screened by the limma (version 3.46.0) R package [13]. The cut-off criteria were set as
Enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) signal pathway
First, methylation-driven genes were converted into IDs by the org.Hs.eg.db (version 3.12.0) R package. GO functional enrichment analysis of the methylation-driven genes was carried out by using clusterProfiler (version 3.18.1) R package (
Construction of a prognostic models of the methylation-driven genes
First, the expression degree and methylation degree of methylation-driven genes were integrated with survival data, and then the linear risk assessment model of the methylation-driven genes was constructed by univariate and multivariate Cox regression analysis with survival R package (
Identification of key coexpression modules by WGCNA
WGCNA mines genes with similar expression patterns based on different gene expression patterns, which are, included in a module. The module features and hub genes within modules were identified, and each module was associated with other modules or external sample features to screen core genes in modules that can serve as biological markers [22]. In this paper, the weighted gene coexpression network of the breast cancer transcriptome dataset was constructed by using the WGCNA package in R software (version 4.0.3) [23]. To further select the functional modules in the coexpression network, we calculated the correlation coefficient between the modules and the phenotype of interest, obtained the correlation degree between the modules and clinical features, and selected the module with a high correlation coefficient for subsequent analysis, which was considered as a functional module related to clinical features.
Methylation-driven genes
Methylation-driven genes
The overlapping genes between the co-expressed genes of functional modules with high correlation coefficients obtained by WGCNA and the prognosis-related methylation-driven genes were regarded as key methylation-driven genes.
Transcriptome and methylation combined survival analysis and expression level verification
To further study the influence of the expression level and methylation degree of the key methylation-driven genes on the prognosis of breast cancer patients, the survival and Survminer R packages were used to analyse the survival rates related in gene expression and methylation, degree of these genes to better predict the prognosis of breast cancer patients [24, 25]. Gene expression profiling interactive analysis (GEPIA) is a public database (
Results
Identification of methylation driven genes in breast cancer
The transcriptome sequencing data of 1222 breast cancer patients were obtained from the TCGA database, including 113 normal patients and 1109 tumour patients. A total of 892 samples had DNA methylation data, including 96 normal samples and 796 tumour samples. With
Multivariate Cox regression analysis of four methylation driven genes
Multivariate Cox regression analysis of four methylation driven genes
Heat map of 17 methylation driver genes. (a) The degree of methylation-driven genes in normal samples and tumour samples. Red indicates that the genes are hypermethylated in tumour and normal samples, green indicates that the genes are hypomethylated in tumours and normal samples. (b) Expression level of methylation-driven genes in normal samples and tumour samples. Red indicates that the genes are upregulated in tumour and normal samples, while green indicates that the genes are downregulated in tumour and normal samples.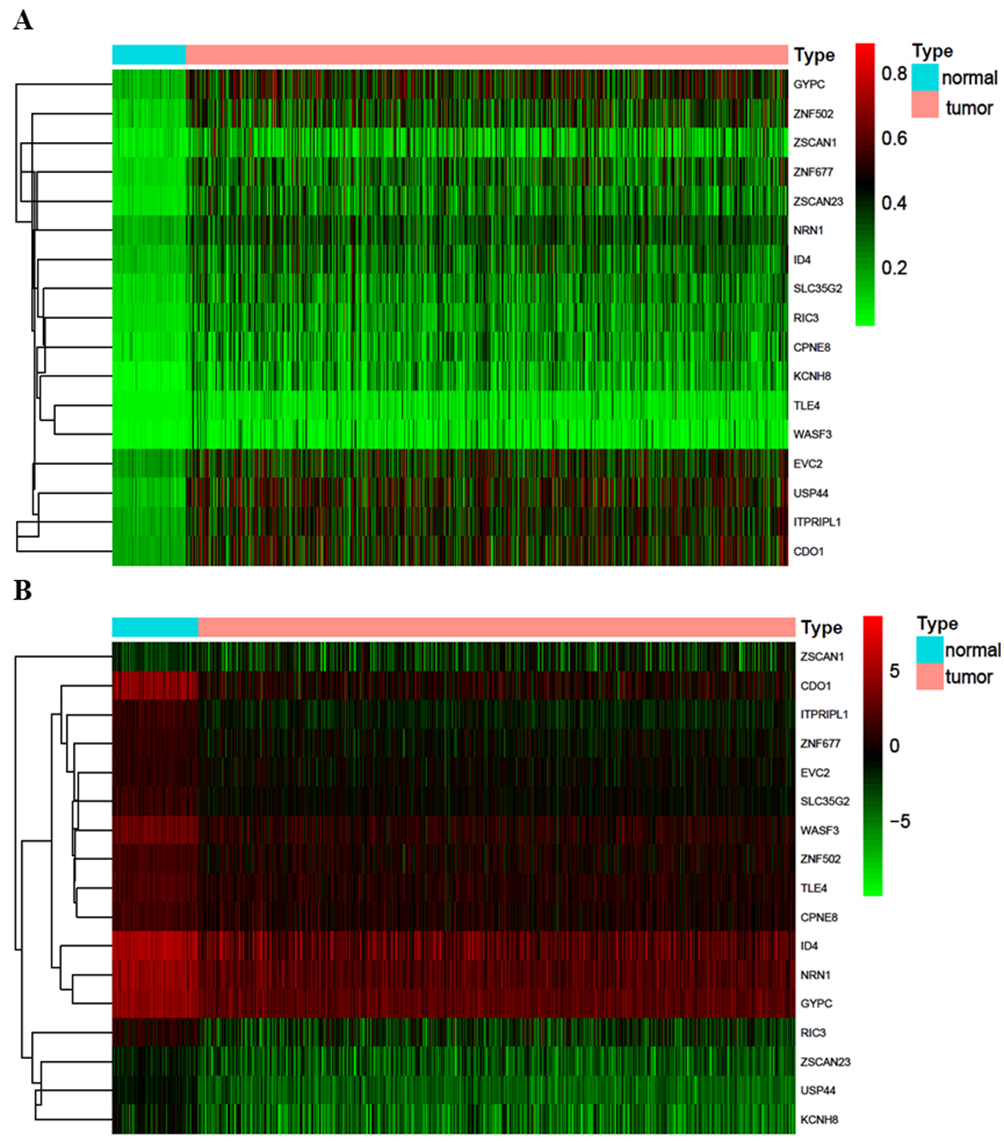
Seventeen methylation-driven genes were analysed in terms of their GO functions and KEGG signalling pathway to identify related biological functions and signalling pathways. The results showed that the methylation-driven genes were mainly enriched in oligodendrocyte development, oligodendrocyte differentiation, glial cell development, myelination, ensheathment of neurons, and axon ensheathment (
Functional enrichment analysis of methylation-driven genes in breast cancer. (a) GO functional enrichment analysis of methylation-driven genes. (b) KEGG signalling pathway analysis of methylation-driven genes.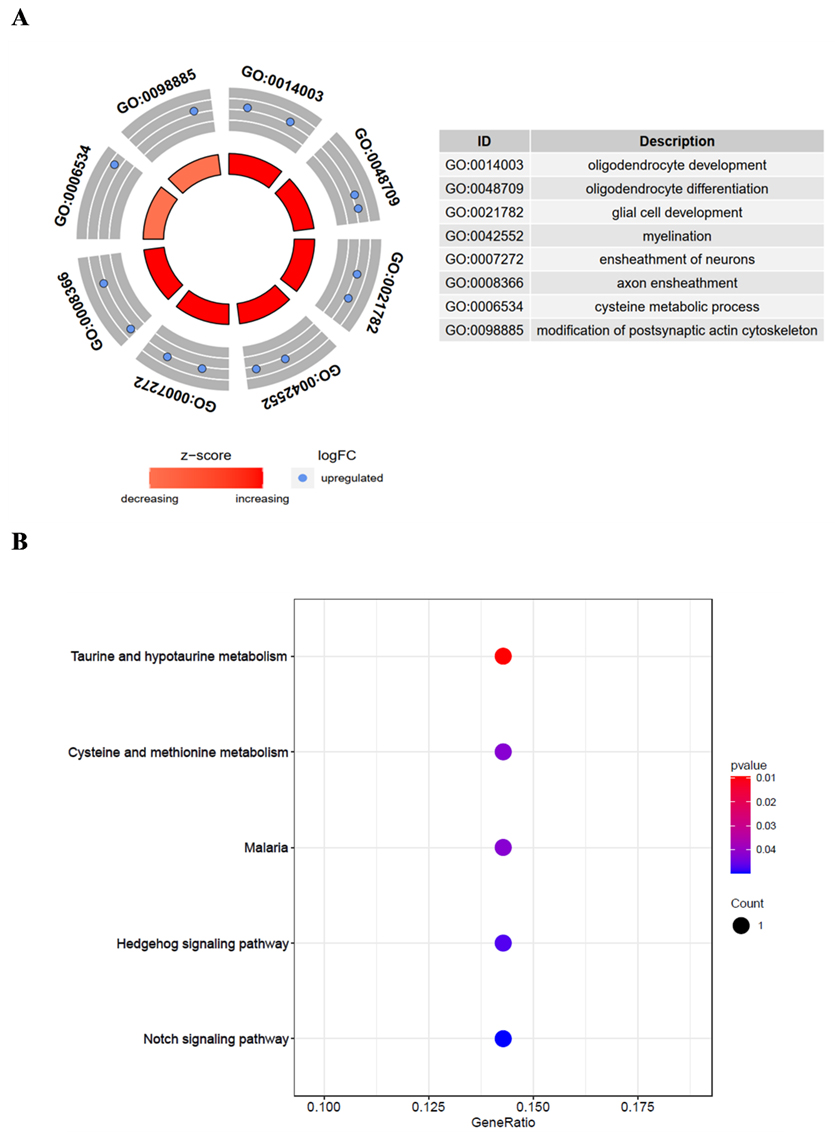
To determine the prognostic role of methylation-driven genes in breast cancer, univariate Cox regression was used to analyse the methylation-driven genes related to prognosis, and 4 risk genes with HR
Prognostic evaluation of the 4 risk models of methylation-driven genes. (a) Survival analysis of the high-risk group and the low-risk group. (b) ROC curve evaluation of the predictive ability of different indicators of methylation driver genes. (c) The distribution of risk scores for breast cancer patients. (d) The survival status of breast cancer patients in the high-risk group and the low-risk group.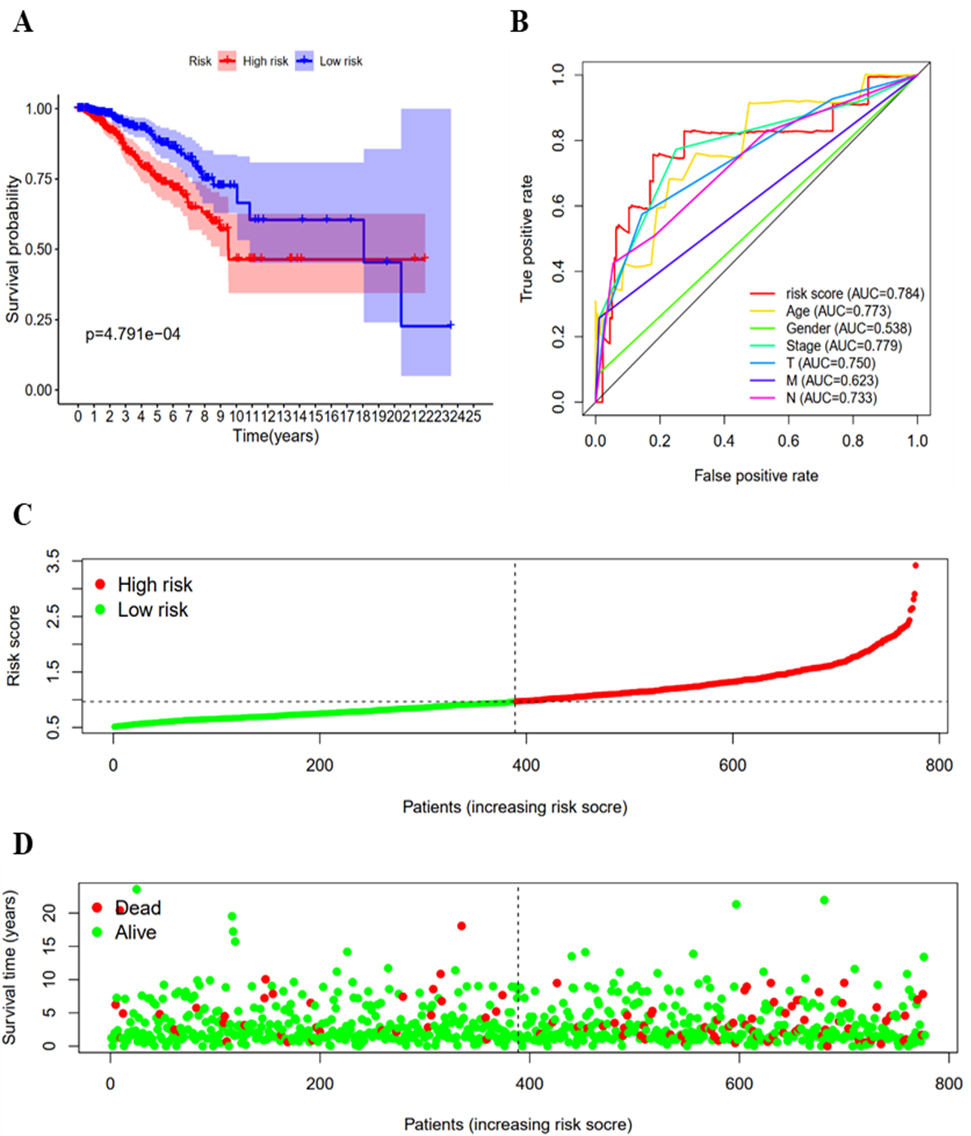
The breast cancer transcriptome data set was analysed by WGCNA. First, sample clustering was performed on the expression matrix, and no outliers were found (Fig. 5A). Second, the clinical data were prepared, imported, and compared with the sample cluster tree. The results showed that the sample cluster tree matched the clinical data (normal and tumour) well (Fig. 5B). Then, the appropriate soft threshold
Weighted gene coexpression network analysis of breast cancer transcriptome data. (a) Cluster analysis of samples. (b) Sample clinical information corresponding to sample clustering. (c) Soft threshold analysis was used to obtain the scale-free fitting index of the network topology. (d) Module clustering tree and module division of coexpressed genes in breast cancer. (e) Correlation between clinic characteristic and trait. The ordinate represents different colour modules, and the abscissa represents clinical information (normal and tumour). The numbers in the module are the correlation and 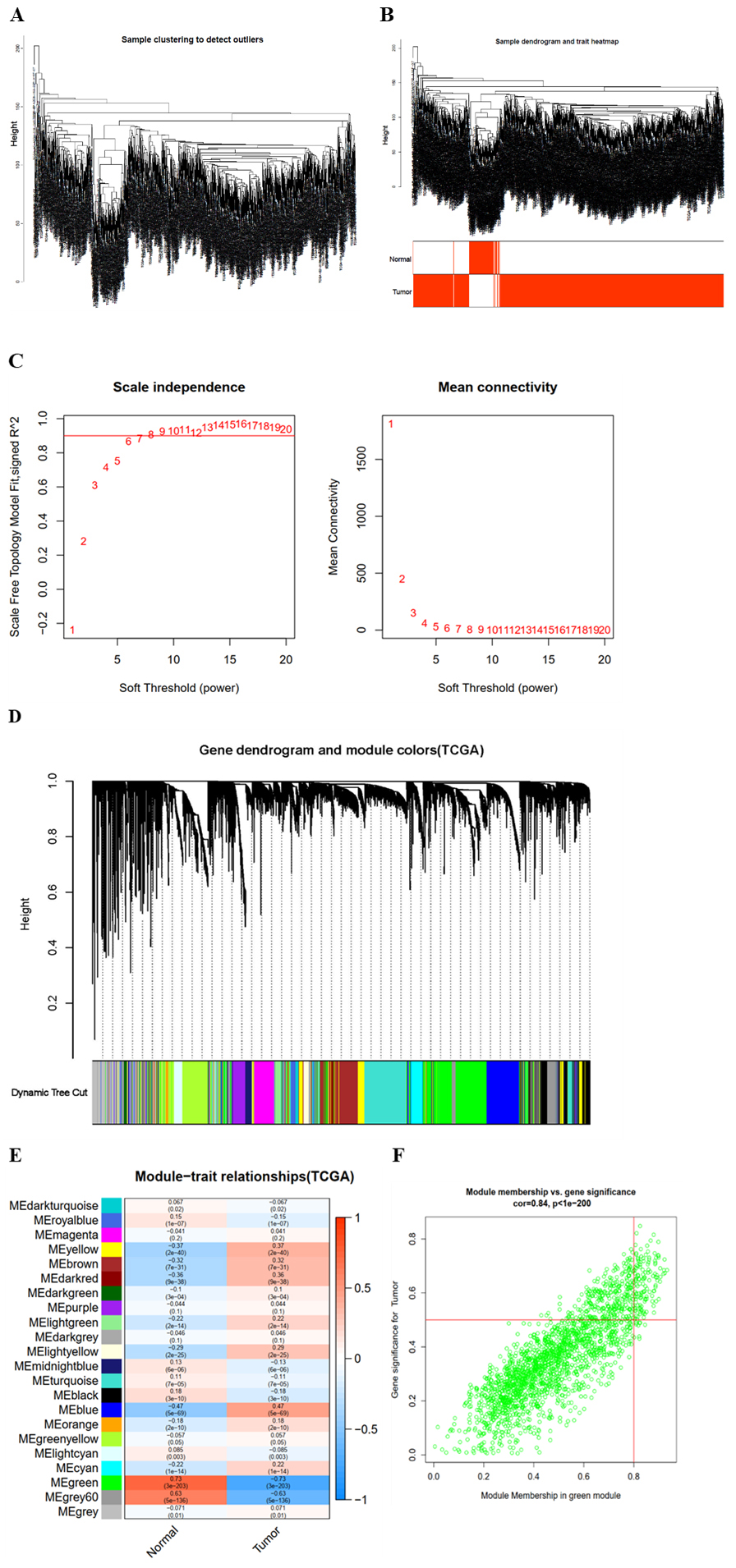
Screening of key methylation-driven genes and analysis of methylation degree. (a) Venn diagram of key methylation-driven genes. (b) Degree of CDO1 in breast cancer and normal tissues. (c) Correlation between the methylation degree of CDO1 and the expression level.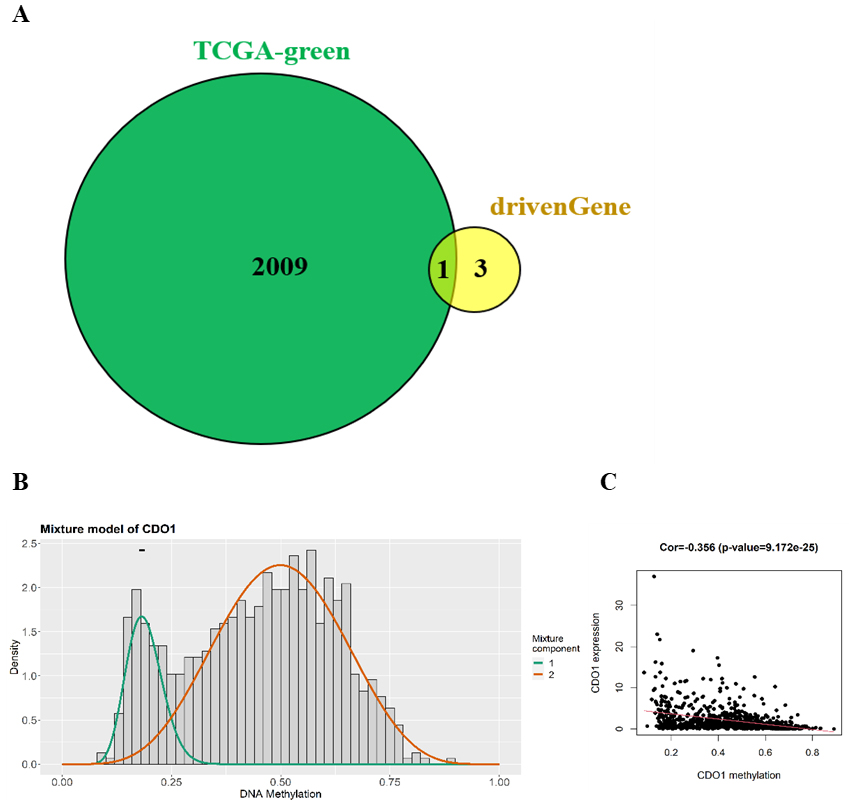
Kaplan Meier combined curve and expression level analysis of CDO1. (a) Combined survival analysis of CDO1 expression and methylation. (b) Expression level of CDO1 in tumour tissues (red) and normal tissues (green). (d) Analysis of the expression level of CDO1 in various tumours.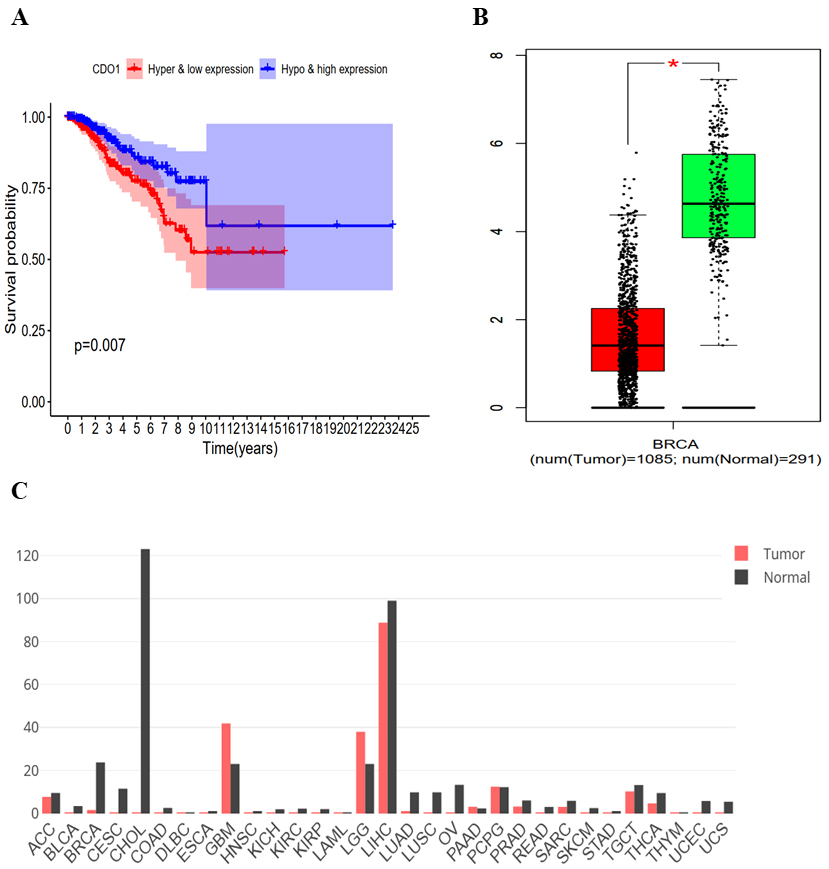
Venn diagram of the 2010 coexpressed genes in the green module and 4 methylation-driven genes related to prognosis was drawn (Fig. 6A). It was found that only CDO1 was an overlapping gene, and it was used as a key methylation-driven gene related to prognosis. The degree of CDO1 methylation in breast cancer patients and normal patients was compared, and the correlation between the degree of methylation and gene expression was determined (Fig. 6B and C). The degree of methylation of the CDO1 gene in tumour samples was higher than that in normal samples, and its methylation degree was negatively correlated with the expression level.
Combined analysis of the methylation and expression of CDO1 and verification of its expression level
Kaplan Meier curve analysis of the methylation-driven genes showed that the combination of the methylation and expression of CDO1 was significantly correlated with the prognosis of breast cancer patients (
Discussion
In our study, we screened 17 methylation-driven genes in breast cancer. Through functional and signalling pathway analysis, it was found that the methylation-driven genes were mainly enriched in oligodendrocyte development, oligodendrocyte differentiation, glial cell development, myelination, ensheathment of neurons, axon ensheathment, taurine and hypotaurine metabolism, cysteine and methionine metabolism, Hedgehog signalling pathway and malaria. Through univariate and multivariate Cox regression analyses, a prognostic model of 5 methylation-driven genes was established. The risk scores of the five methylation-driven genes were calculated, and the breast cancer patients were divided into a high-risk groups (
The combined survival analysis of methylation-driven gene expression and methylation showed that the survival rate of patients with hypermethylation and low CDO1 gene expression was lower.
CDO1 (cysteine dioxygenase type 1) is a tumour suppressor gene, and its encoded protein is non-heme ferrase, which is involved in the process of converting cysteine into cysteine sulfinic acid affecting mitochondrial function, CDO1 also inhibits the production of glutathione in cysteine, which leads to an increase in reactive oxygen species and promotes cell apoptosis [28, 29]. Hypermethylation of CDO1 was highly specific in human cancers and has been reported as a prognostic marker in many cancers [30]. Harada et al. found that the prognosis of gastric cancer patients with hypomethylation of the CDO1 gene was significantly higher than that of gastric cancer patients with hypermethylation of the CDO1 gene [31]. Kojima et al. found that the prognosis of oesophageal cancer patients with CDO1 gene methylation was worse than that of oesophageal cancer patients without CDO1 gene methylation [32]. In lung cancer, promoter methylation of CDO1 can be used as a prognostic marker [33]. Liew et al. found that the CDO1 gene can be used as a good epigenetic biomarker in endometrial carcinoma [34]. More importantly, it has been reported that the prognosis of breast cancer patients with hypermethylation of CDO1 was significantly lower than that of patients with hypomethylation of CDO1, and DNA methylation of the CDO1 promoter can be used as a reliable prognostic indicator for primary breast cancer patients who have not received chemotherapy [35]. According to our research, the expression level of the CDO1 gene was negatively correlated with the degree of methylation in breast cancer, and the prognosis of breast cancer patients with low expression levels and high methylation levels of CDO1 was poor. Consistent with our prediction, the CDO1 gene is a tumour suppressor gene. We suspect that the promoter region of the CDO1 gene is hypermethylated, which will down-regulate or hinder the expression of CDO1 gene. Therefore, we predict that the hypermethylation of the CDO1 gene can be an important indicator of the prognosis of breast cancer patients.
In summary, the key methylation-driven gene CDO1 was screened based on WGCNA has a certain diagnostic value for breast cancer and can be used as a potential biomarker for predicting the prognosis of breast cancer.
Author contributions
Conception: Simei Tu.
Interpretation or analysis of data: Simei Tu, Hao Zhang.
Reparation of the manuscript: Simei Tu, Xinjian Qu.
Revision for important intellectual content: Simei Tu, Xinjian Qu.
Supervision: Xinjian Qu.
Footnotes
Acknowledgments
Not applicable.