
Abstract
Breast cancer (BC) is an aggressive cancer with a high percentage recurrence and metastasis. As one of the most common distant metastasis organ in BC, lung metastasis has a worse prognosis than that of liver and bone. Therefore, it’s important to explore some potential prognostic markers associated with the lung metastasis in BC for preventive treatment. In this study, transcriptomic data and clinical information of BC patients were downloaded from The Cancer Genome Atlas (TCGA) database. Co-expression modules constructed by weighted gene co-expression network analysis (WGCNA) found the royal blue module was significantly associated with lung metastasis in BC. Then, co-expression genes of this module were analyzed for functional enrichment. Furthermore, the prognostic value of these genes was assessed by GEPIA Database and Kaplan-Meier Plotter. Results showed that the hub genes, LMNB and CDC20, were up-regulated in BC and had a worse survival of the patients. Therefore, we speculate that these two genes play crucial roles in the process of lung metastasis in BC, which can be used as potential prognostic markers in lung metastasis of BC. Collectively, our study identified two potential key genes in the lung metastasis of BC, which might be applied as the prognostic markers of the precise treatment in breast cancer with lung metastasis.
Introduction
Breast cancer (BC) is the most common malignancy in the female population, accounting for about 30% of all female cancers [1]. Though the diagnosis and treatment methods have developed rapidly in recent decades, the mortality of BC still remains high due to the frequent distal metastasis [2, 3, 4]. The metastases at distant sites are the main cause of breast cancer death, therefore, the presence and location of breast cancer metastasis have been the critical diagnose to the clinical course and prognosis of patients [5, 6]. Nevertheless, the prognosis of distant metastasis is significantly affected by the site of initial spread [7]. Among the priority sites of breast cancer transmission, metastasis to the lung has a worse prognosis than those to the liver or bone [8]. This is mainly due to little or no symptoms of early lung metastasis, and large metastases and severe symptoms have been caused when found the breast cancer lung metastases [9]. However, to date, there is no effective treatment for different metastatic sites, especially for the lung metastasis.
Different primary tumors have a proclivity to metastasize to distinct organs. The “seed” and “soil” theory has put forward that the distant metastasis of tumor was not accidental, but a certain biocompatibility existed between tumor cells and target organs, and tumor metastasis was affected by the effect of driver genes and target organ microenvironment [10]. However, identifying the specific driver genes is still challenging.
WGCNA is a systematic biological method which can analyze multiple gene expression patterns in multiple samples [11]. Through constructing gene co-expression modules based on their expression patterns, WGCNA can analyze the relationship between modules and specific phenotypes, for example the clinical symptoms of patients. Comparing with traditional molecular biology, the unique advantage of WGCNA is that it can convert gene expression data into co-expression modules, and provide insight into the signaling networks responsible for phenotypic traits of interest. It has been successfully used to study various biological processes, such as gastric cancer [12], colon adenocarcinoma [13], liver hepatocellular carcinoma [14] and glioma [15], proving its effective to identify the potential biomarkers and therapeutic targets. It not only helps to compare the expression of different genes, but also helps to calculate and analyze the interactions between genes in different co-expression modules. WGCNA delivers a more comprehensive exploration of the whole biological system in diseases and will be quite helpful to identify the candidate biomarkers or therapeutic targets.
Here we used WGCNA to explore the relationship between gene expression and metastatic site of breast cancer. We find out the meaningful module correlated with lung metastasis of breast cancer. GO enrichment and KEGG pathway analysis were performed to figure out the main functions of genes in the main module. Cox regression analysis was performed to pick out the hub genes of the main module. GEPIA and Kaplan-Meier Plotter were used to identify the expression value and prognostic value of these genes. We believe that our work will lay an important foundation for the future treatment of lung metastasis of breast cancer and improve the overall survival rate of breast cancer.
Material and methods
Patient samples data collection and processing
Public gene-expression data and clinical annotation were downloaded from the cBioportal online database (
Weighted gene co-expression network construction
The Flash Clust tools package was used to perform the cluster analysis. In the present study, the soft-threshold
Clinically significant modules identification
The co-expression module is defined as a class of genes with high topological overlap similarity, and genes in the same module generally have a higher degree of co-expression. In this study, two methods were used to identify the important modules associated with clinical traits. First, the module eigengene (ME) represents the principal component of the module to describe the expression pattern of the module in each sample. Second, module membership (MM) refers to the correlation coefficient between genes and module eigengenes to describe the reliability of a gene belonging to a module. Finally, the correlation was calculated between the modules and the clinical data to identify significantly clinical modules.
Identification and validation of Hub Genes
The Cytoscape software (version 3.7.1) was used to determine the Hub Genes. The module membership can be ascertained by calculated the intra-modular connectivity. In this study, 0.1 were set as the cut-off criteria to screen out hub genes.
Gene ontology enrichment and KEGG pathway analysis
WebGestalt (
The number of genes in the 39 module
The number of genes in the 39 module
Multivariate Cox regression was performed using SPSS software (IBM SPSS Statistics 22). Thirty-nine genes were selected for screening the optimal prognostic signatures for breast cancer with lung metastasis. All data was evaluated by the Pearson’s Chi-Square method with SSPS software (IBM SPSS Statistics 22).
GEPIA database
GEPIA (
Kaplan-Meier plotter analysis
In our study, we used Kaplan-Meier plotter (
bc-GenExMiner v4.0
Breast Cancer Gene-Expression Miner v4.0 (
Results
Construction of co-expression modules of breast cancer
The flowchart identifying the multi-gene signatures with breast cancer lung metastasis.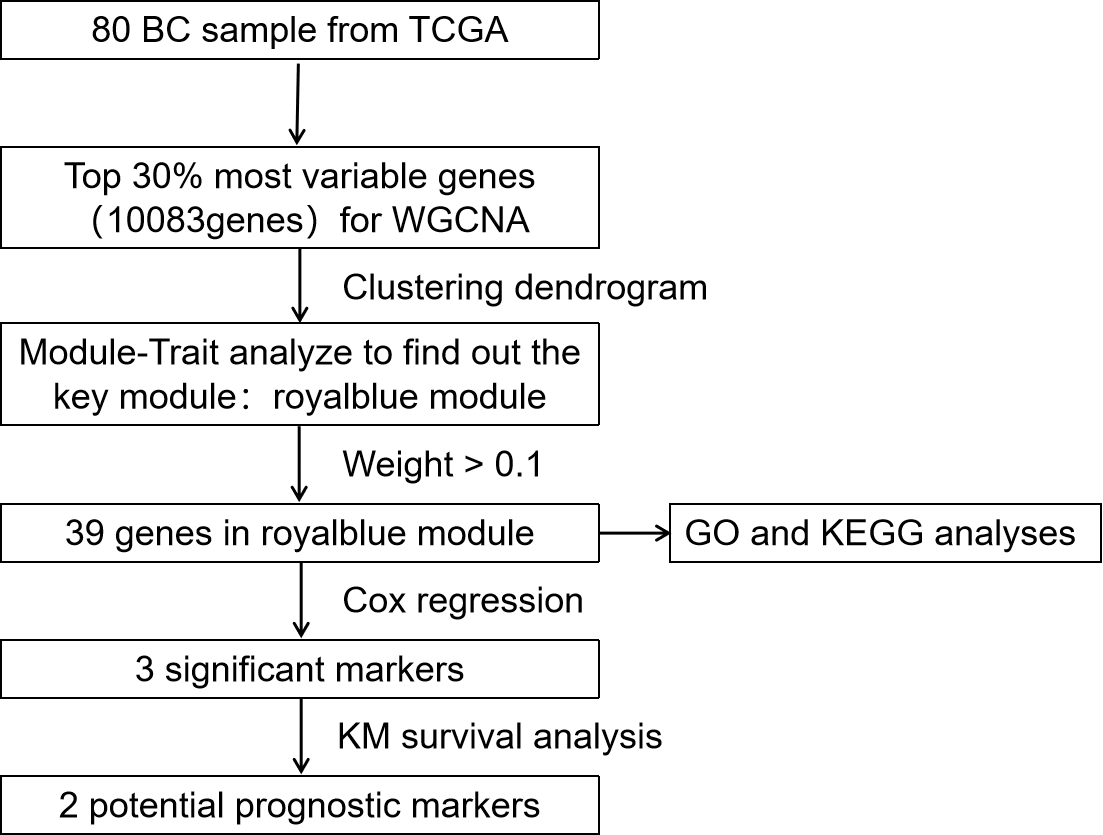
Determination of soft-threshold power in the WGCNA. (A) Clustering tree based on the module eigengenes of modules to detect outliers. (B) Scale-free index for various soft-threshold powers. (C) Mean connectivity for various soft-threshold powers.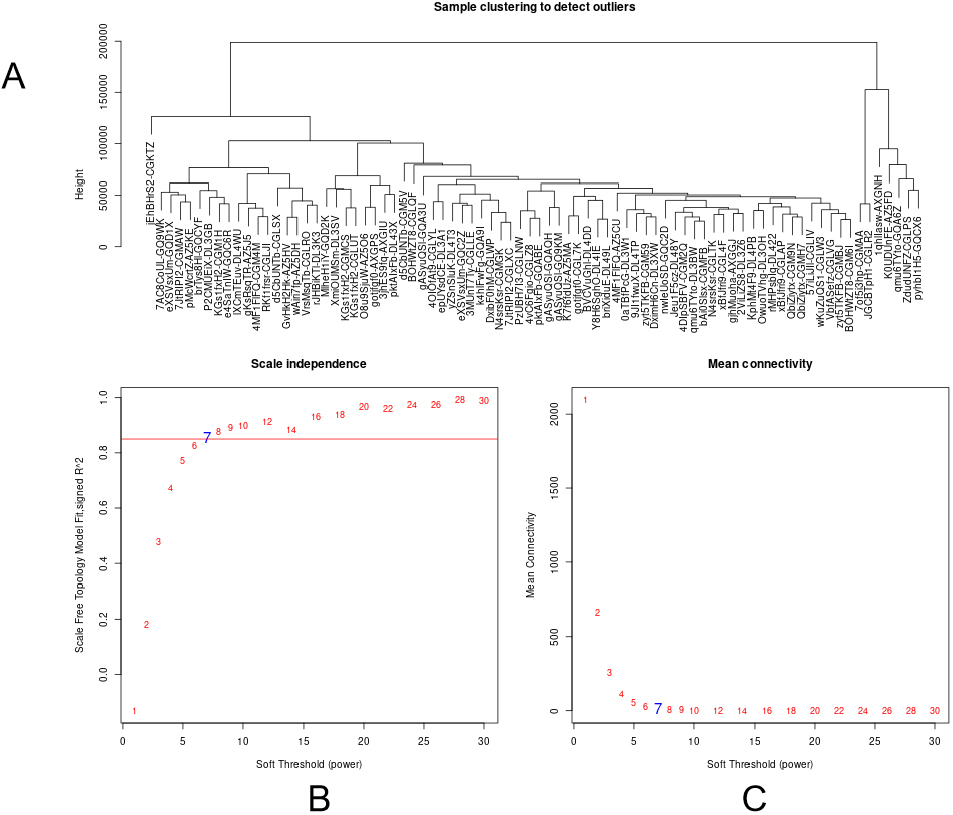
Clustering dendrograms of genes, with dissimilarity based on topological overlap, together with assigned module colors. As a result, 39 co-expression modules were constructed and was shown in different color. These modules were ranged from large to small by the number of genes they included. The number of genes in the 39 modules was listed in Table 1.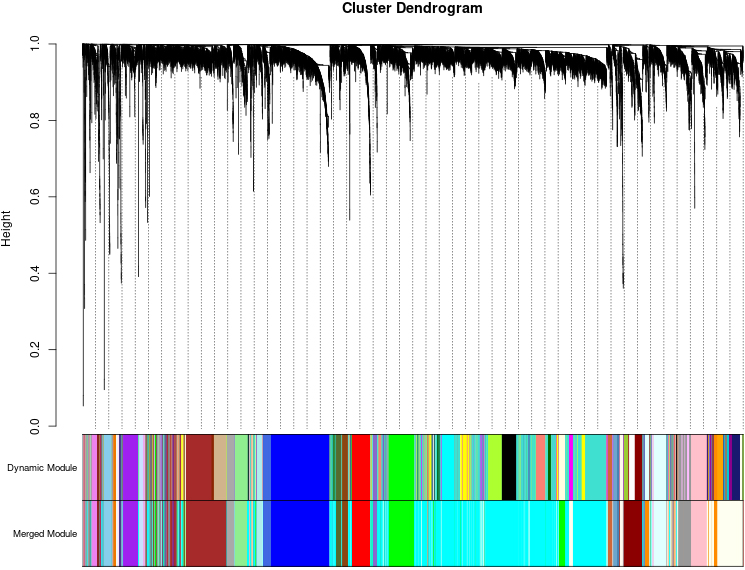
Visualizing the gene network using a heatmap plot. The heatmap depicts the Topological Overlap Matrix (TOM) among all genes in the analysis. Light color represents low overlap and progressively darker red color represents higher overlap. Blocks of darker colors along the diagonal are the modules. The gene dendrogram and module assignment are also shown along the left side and the top.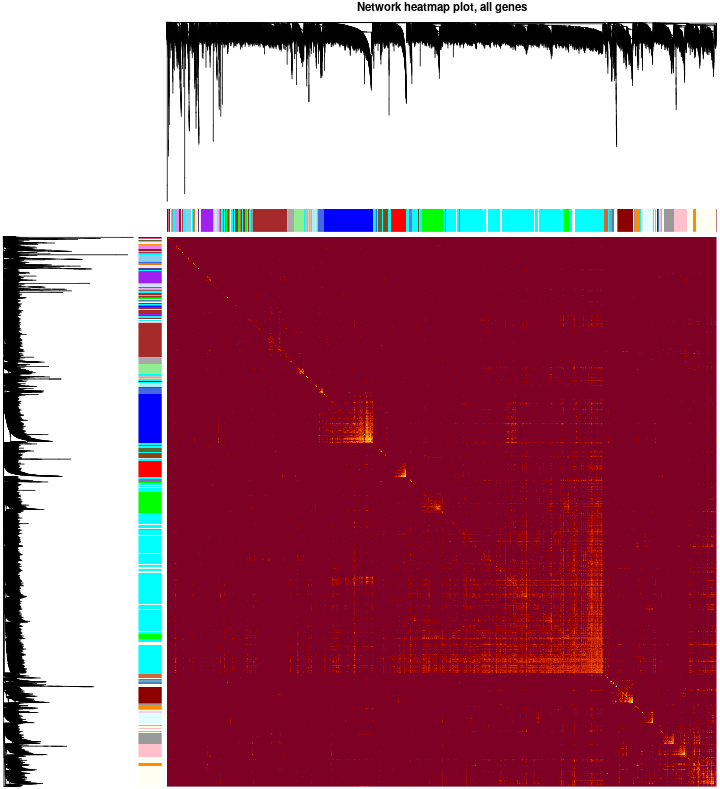
The data was processed and analyzed following the flowchart (Fig. 1). Expression values of top 30% most variable genes (10083 genes) of breast cancer were used to construct the co-expression module by WGCNA package tool. The cluster analysis on these samples were performed by the Flash Clust tools package and the result was shown in (Fig. 2A). The power value, which mainly affected the independence and the average connectivity degree of co-expression modules, was screened out and equal to seven (Fig. 2B) and the independence degree was up to 0.8. Therefore, the power value was used to build gene co-expression module and the results showed that there were 39 modules in breast cancer. These co-expression modules were displayed in different colours (Fig. 3). These modules were ranged from large to small by the number of genes they included. The number of genes in the 39 modules was shown in (Table 1). Interactions of the 39 co-expression modules were analyzed and shown in (Fig. 4).
The clinical information was provided by TCGA database. And we selected the metastasis organs as research targets and removed the patient sample information which was meaningless or lacking in our study. According to the correlation between module eigengene and clinic traits, the interaction of co-expression modules and particular traits were identified (Fig. 5). We found that the royalblue module (
Module-trait associations. Each row corresponds to a module eigengene, column to a trait. Each cell contains the corresponding correlation and 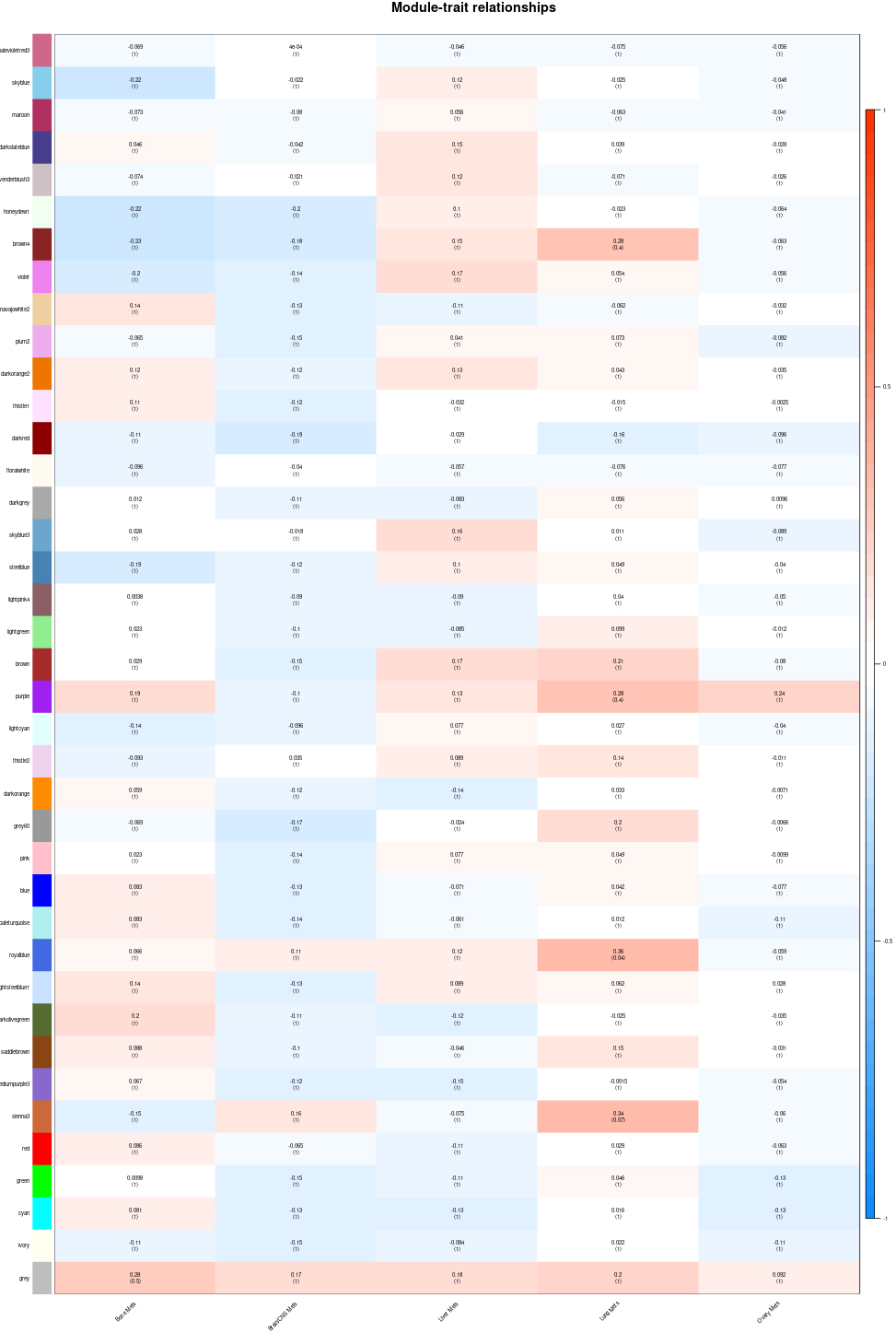
GO enrichment analysis and KEGG analysis were performed on the genes in the royal blue module. The biological process of most genes was metabolic process, biological regulation and cellular component organization (Fig. 7). The cellular component of most genes was nucleus, and the molecular function of most genes was protein binding (Fig. 7). According to KEGG analysis, genes in royal blue module were mainly enriched in systemic lupus erythematosus, reproduction, alcoholism, RHO GTPase effectors, viral carcinogenesis, signaling by Rho GTPase, cell cycle, developmental biology, generic transcription pathway (Fig. 8).
The multivariate cox regression analysis between markers and lung metastasis status
The multivariate cox regression analysis between markers and lung metastasis status
Omnibus test of model coefficients
Method
The eigengene dendrogram and heatmap identify groups of correlated eigengenes termed meta-modules.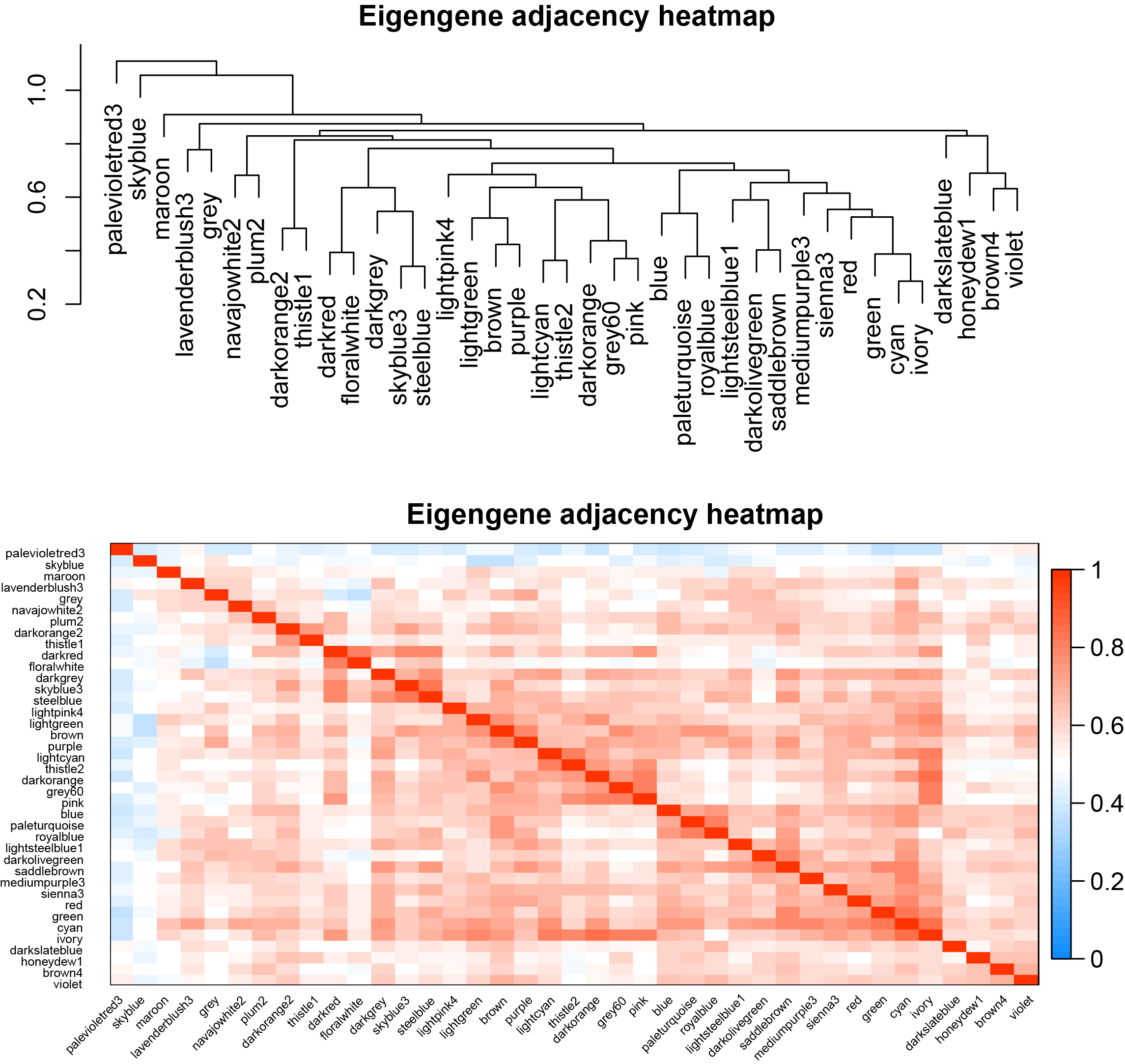
GO analyses. Bar plot represent the biological process, cellular component and molecular function of hub genes in the royal blue module.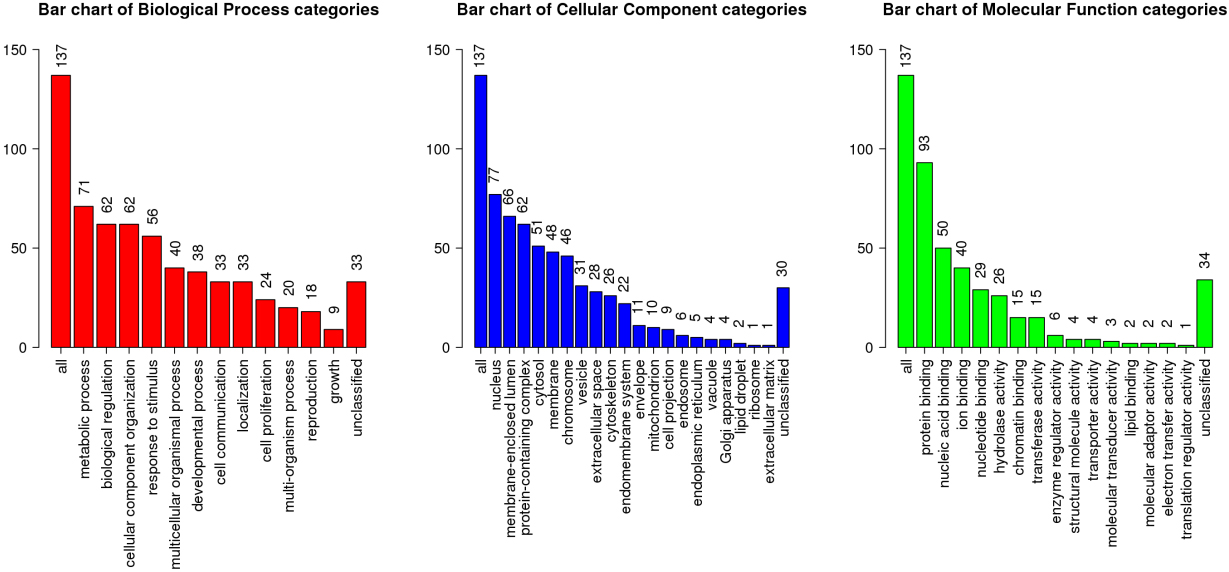
KEGG analyzes. Bar plot represent the pathways of the hub genes in the royal blue module.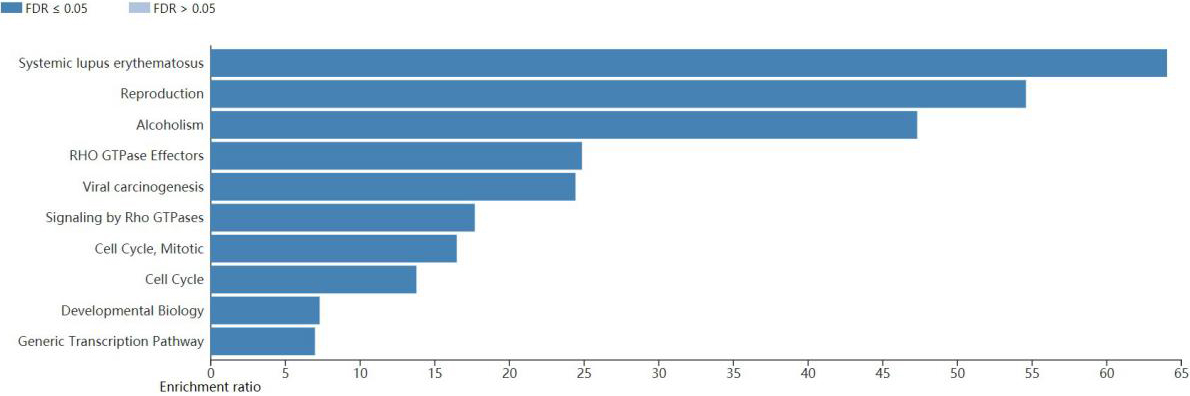
Construction of PPI network of genes in royal blue module. The PPI network of hub genes was analyzed by Cytoscape software.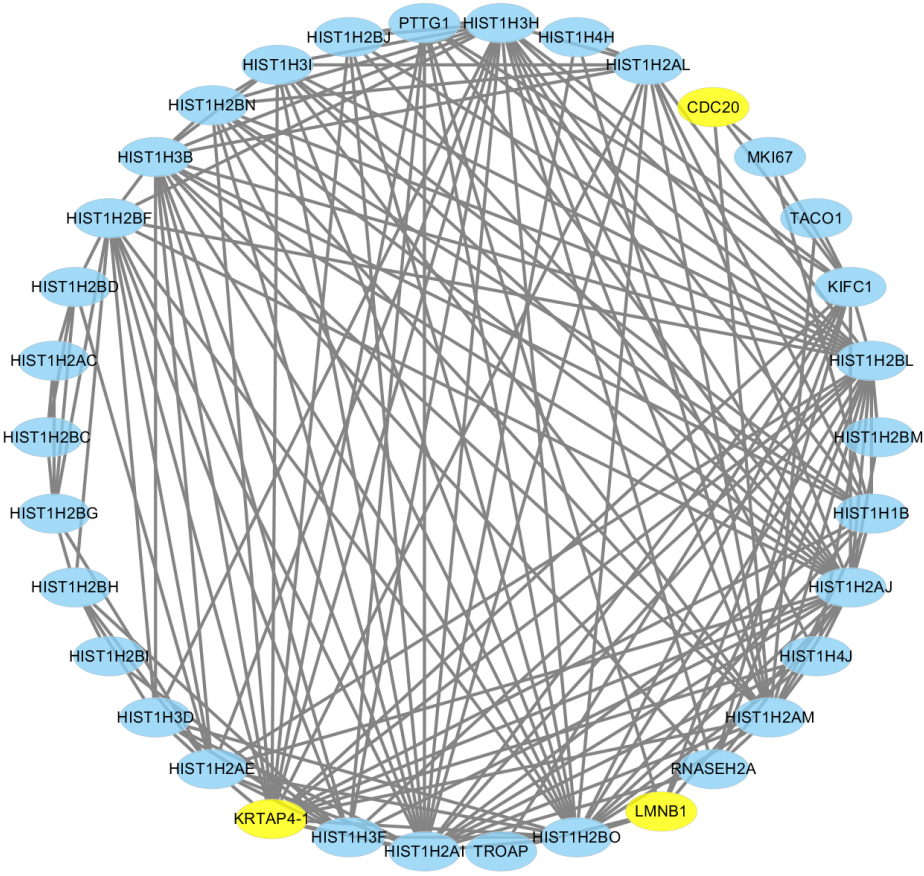
Verify the prognostic significance of hub genes in breast cancer patients. A. The mRNA expression of LMNB1 and CDC20 in breast cancer tissues was analyzed using GEPIA database (BRCA) (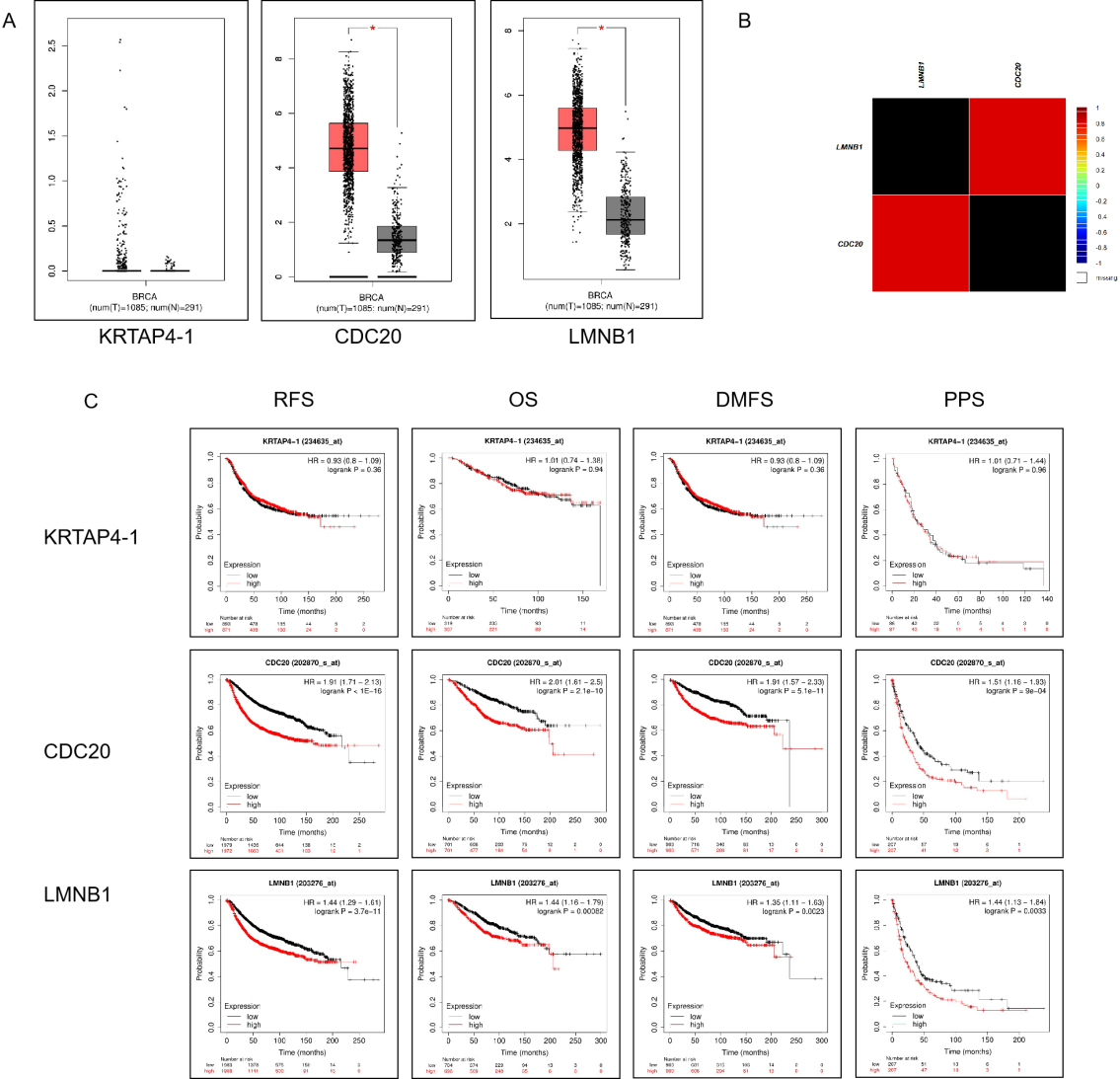
The genes in royal blue module were calculated the intramodular connectivity. The intramodular connectivity was calculated for each gene by summing the connection strengths with other module genes and dividing this number by the maximum intramodular connectivity. Total of 39 genes were connectivity greater than 0.1, and then these genes were selected as hub genes and then analyzed using the Cytoscape software (Fig. 9). Next, we integrated the expression profiles of 39 genes in the module with the occurrence of pulmonary metastasis in the corresponding 80 patients and conducted Cox regression analysis. Finally, KRTAP4-1, LMNB1 and CDC20 were independent risk factors for breast cancer with lung metastasis. The relative risk between these three genes and lung metastasis of breast cancer were 1.146, 1.269 and 0.885, respectively (Table 2). The omnibus test of model coefficients showed that the overall test of the model was statistically significant (Table 3). And then we found LMNB1 and CDC20 were up-regulated in breast cancer tissue through the GEPIA database. Moreover, we also found that the over-expression of this two genes predict worse survival of breast cancer using the Kaplan-Meier plotter website (Fig. 10). We further verified the correlation between LMNB1 and CDC20 using the bc-GenExMiner v4.0, the result showed that the correlation coefficient was up to 0.9.
Discussion
Lung metastasis is a pernicious outcome of breast cancer. About 17% of patients with BC have a propensity developing to lung metastases [16]. Based on the selective evolution of organs, metastatic localization does not occur randomly, but prefers a location controlled by numerous micro-environmental, cellular, and molecular factors [9]. Understanding the mechanism of lung metastasis of breast cancer is helpful for further prevention and even targeted therapy. There are many theories to explain the process of lung metastasis in breast cancer: for example, the interaction between CSCs forming breast cancer cells and pulmonary vessels [17, 18, 19], the histologic and intrinsic genomic profiles of breast cancer [20], barrier drive of host organs [21]. Individual genes are associated with organ-specific metastasis. Multiple individual genes constitute a complex, dynamic and interactive network and govern the process of breast cancer cell metastasis to specific organs [4, 22]. To date, Many bioinformatics studies have found gene clusters associated with poor prognosis in breast cancer [23], but whether they are associated with organ-specific metastasis still not clear. No studies have correlated gene expression with clinical traits and used it to predict lung metastasis of breast cancer. Our result indicated that CDC20 and LMNB1 were co-expressed in breast cancer and played important roles in the process of lung metastasis in breast cancer, suggesting their important value for further study.
LMNB1 is a nuclear membrane protein that can build a framework for the nuclear envelope [24]. LMNB1 is also essential for cell senescence [25]. For example, down-regulation of LMNB1 induces cellular senescence through activating either the p53 or pRB tumor suppressor pathway [26]. Increasing evidence shows that up-regulation or down-regulation of LMNB1 affects the clinical behavior of cancer. Furthermore, previous study also showed LMNB1 is associated with lung metastasis in breast cancer in mice [27]. Therefore, we conclude that LMNB1 plays an important role in the process of lung metastasis in breast cancer, whereas further confirming evidence of this result in human breast cancer is still need.
CDC20 is a spindle assembly checkpoint molecule, which is critical in cell cycle progression [28]. Aberrant expression of CDC20 is associated with malignant progression and poor prognosis in various types of cancer, such as astrocytoma [29], gastric cancer [30], hepatocellular carcinoma [31, 32], colorectal cancer [33, 34] and non-small cell lung cancer [35]. In addition, there is a significant correlation between a high expression of CDC20 and advanced tumor stage in carcinoma [36]. These studies showed that over-expression of CDC20 were related with cancer metastasis and poor prognosis. Song et al. had found that CDC20 were significantly up-regulated in triple negative breast cancer (TNBC) and correlated with tumor formation and metastasis of TNBC [37], indicating the prognosis value of CDC20 in breast cancer. Our study drew further conclusions. To the best of our knowledge, our study is the first time to further verify that CDC20 is related with the progress of lung metastasis in breast cancer, indicating that CDC20 can be a prognostic marker or a potential therapy target for breast cancer with lung metastasis.
In conclusion, our study is the first study to screen the characteristic hub genes and construct a prognostic model based on hub genes in breast cancer with lung metastasis using WGCNA. The potential prognostic predictive genes, LMNB1 and CDC20, have been proved to be able to investigate the occurrence of lung metastasis in breast cancer. Therefore, these two genes might be used as the potential biomarkers to identify the high-risk patients and assess the prognosis to facilitate the precise treatment in breast cancer with lung metastasis.
Authors contributions
X.X. analyzed the data, and wrote and revised the manuscript.
Declaration of interest statement
The author declares no conflicts of interest with the contents of this article.
Footnotes
Acknowledgments
This study was supported by the Youth Program of National Natural Science Foundation of China (81802956).