
Abstract
Objective:
Invasive breast cancer (BRCA) is one of the prevalent types of invasive tumors with high mortality worldwide. Due to the lack of effective treatment to control the recurrence of distant metastases, the prognosis of BRCA is still very unsatisfactory. We aimed to find some biomarkers by bioinformatics analysis for survival prediction.
Methods:
Differentially expressed genes (DEGs) were screened out based on tumor group and normal group. Then, the weighted gene correlation network analysis (WGCNA) was employed to identify the clinically associated gene sets. Meanwhile, the enrichment analyses were performed for the functional annotation of the critical genes. The Kaplan Meier analysis calculated the essential genes’ prognostic value.
Results:
After threshold screening, 1655 DEGs were obtained for subsequent analysis. 51 out of 1655 DEGs were significantly associated with BRCA patients’ estrogen receptor status via WGCNA. Three genes (FABP7, CXCL3, and LOC284578) out of the 51 genes were associated with overall survival, and 3 genes were relapse-free survival associated. Finally, we obtained 5 essential prognostic associated genes (FABP7, CXCL3, LOC284578, CAPN6, and NRG2), which could be used as prognostic factors for BRCA.
Conclusion:
Our findings obtained a gene module associated with BRCA clinical trait and several key genes that acted as essential components in the prognostic of cancer, which may improve its treatment.
Introduction
Breast cancer is one of the malignant tumors with high mortality worldwide. 1 It is a heterogeneous disease with different molecular subtypes, cell content, clinical manifestation, and treatment response. 2,3 According to reports, after breast protection therapy (BCT) for invasive breast cancer (BRCA), the patient’s treatment and pathological factors are associated with an increased risk of recurrence of ipsilateral breast tumors. 4,5 It is estimated that there will be 276,480 new cases and 42,170 deaths from BRCA in the United States in 2020. 6 At present, BRCA treatment mainly includes surgery, radiotherapy, chemotherapy, and endocrine therapy. 7,8 However, due to the lack of effective treatment to control the recurrence of distant metastases, especially in advanced patients, the prognosis of BRCA is still very unsatisfactory. 9 In recent years, biologically targeted therapy has been proven to be useful for various cancers, significantly improving the prognosis. 10 -13 Therefore, the identification of new biomarkers is essential for the specific treatment of patients.
WGCNA is based entirely on scale-free networks and is used to determine the relationship between genes, thereby identifying modules (clusters) of highly related genes. 14 WGCNA is an ideal method for identifying gene modules and determining essential genes for phenotypic traits. For example, Zou et al. used WGCNA to detected the loss of MAGI2 promotes chronic kidney disease, which regulates cytoskeletal rearrangement in podocytes. 15 Liang W et al. also used WGCNA to identified several key genes that acted as essential components of diabetes-associated cardiovascular disease. 16 Therefore, in this study, our goal is to use the WGCNA algorithm to identify highly relevant gene modules related to breast cancer development and then detect the hub gene (network center gene) to discover new proven effective breast cancer diagnosis and treatment biomarkers.
Methods
Data Sources
The high-throughput RNA-seq data of 1241 patients with BRCA and 113 normal samples were downloaded from the TCGA database. The gene expression profiles were quantified by fragments per kilobase of transcript per million mapped reads (FPKM) normalized estimation and log2-based transformation. We selected 838 BRCA patients with overall survival and relapse-free survival information for further analysis. The following corresponding clinical characteristics were also extracted from the TGGA.
Differential Analysis
The differential expression analyses were conducted between 838 BRCA patients and 113 normal samples using the “limma” R package. 17 In this study, genes with an absolute log2 fold change > 0.585, and adjusted p-value < 0.05 are differentially expressed.
Co-Expression Module Detection
We used the WGCNA R package to construct the co-expression network. At first, a network construction function was used to construct the co-expression network of all the differentially expressed genes (DEGs). The “pickSoftThreshold” R function was used to calculate the soft threshold power, and the “softConnectivity” function was used to calculate the network’s scale-free value. Second, hierarchical clustering and the dynamic tree cut function were used to detect modules. Next, all modules were related to clinical information by “cor” function based on correlation analysis. Gene significance (GS) and module membership (MM) were calculated to relate modules to clinical traits. Finally, the key genes from the preserved module were explored. The correlation (cor.) Gene GS > 0.2 and cor. Gene MM > 0.7 was the inclusive criterion for screening key genes. 18
Function Enrichment Analysis
For the DEGs, KEGG pathway analysis 19 and GO enrichment analysis 20 was performed using Metascape (http://metascape.org) website tool to explore the potential function. 21 Only the tasks with an adjust-p-value <0.05 were selected.
Survival Analysis
Kaplan-Meier analysis with log-rank test was conducted by “survival” R package to screen prognostic associated vital genes. Survival curves were drawn to illustrate the associations of expression levels of these genes with BRCA. The OSbrca Tool was used to testify the survival ability of potential prognostic biomarkers. 22
Statistical Analysis
All analyses were conducted using R software (version 3.6.3) with related packages. T-test was conducted to compare the differences. And adjusted p-value < 0.05 was regarded as significant.
Results
Identification of Differentially Expressed Genes in BRCA
We downloaded transcriptome and clinical data of 838BRCA cases from the TCGA database. Based on our threshold value (absolute log2 fold change > 0.585 and adjusted p-value <0.05), there were 1655 DEGs between BRCA patients and normal groups (Figure 1A). Then, the function analysis showed that these DEGs involved in the IL-17 signaling pathway, PPAR signaling pathway and cell adhesion molecules (CAMs), and so on, and they also associated with receptor regulator activity and growth factor activity, etc (Figure 1B&C). It showed that these DEGs might play a cure role in invasive breast carcinoma.

Differential Analysis of TCGA BRCA samples.
Construction and Analysis of Gene Co-Expression Modules
After differential analyses, a co-expression network of 1655 DEGs were conducted (Figure 2). We first calculated the soft threshold power, which could help us construct a more suitable system based on the co-expression similarity. The function “pickSoftThreshold” in the WGCNA package was used to perform the analysis of network topology. We constructed the gene network and identified modules using the 1-step network construction function. To cluster splitting, the minimum module size was set at 30. The soft threshold power was set at 5 in the subsequent analysis because the scale independence reached 0.85 (Figure 2A and B). The soft power also can assure a scale-free topology model (scale-free R2 = -0.95, Figure 2C). Finally, 5 gene co-expression modules (turquoise, blue, yellow, brown, and green) were constructed (Figure 2D). We analyzed the connectivity of eigengenes. Eigengenes can provide information about the relationship between the gene co-expression modules. The results showed that 5 modules could be clustered into 2 clusters (Figure 2E). Four combinations (modules turquoise, modules blue, modules yellow, and modules brown and green) had a high degree of interaction connectivity.
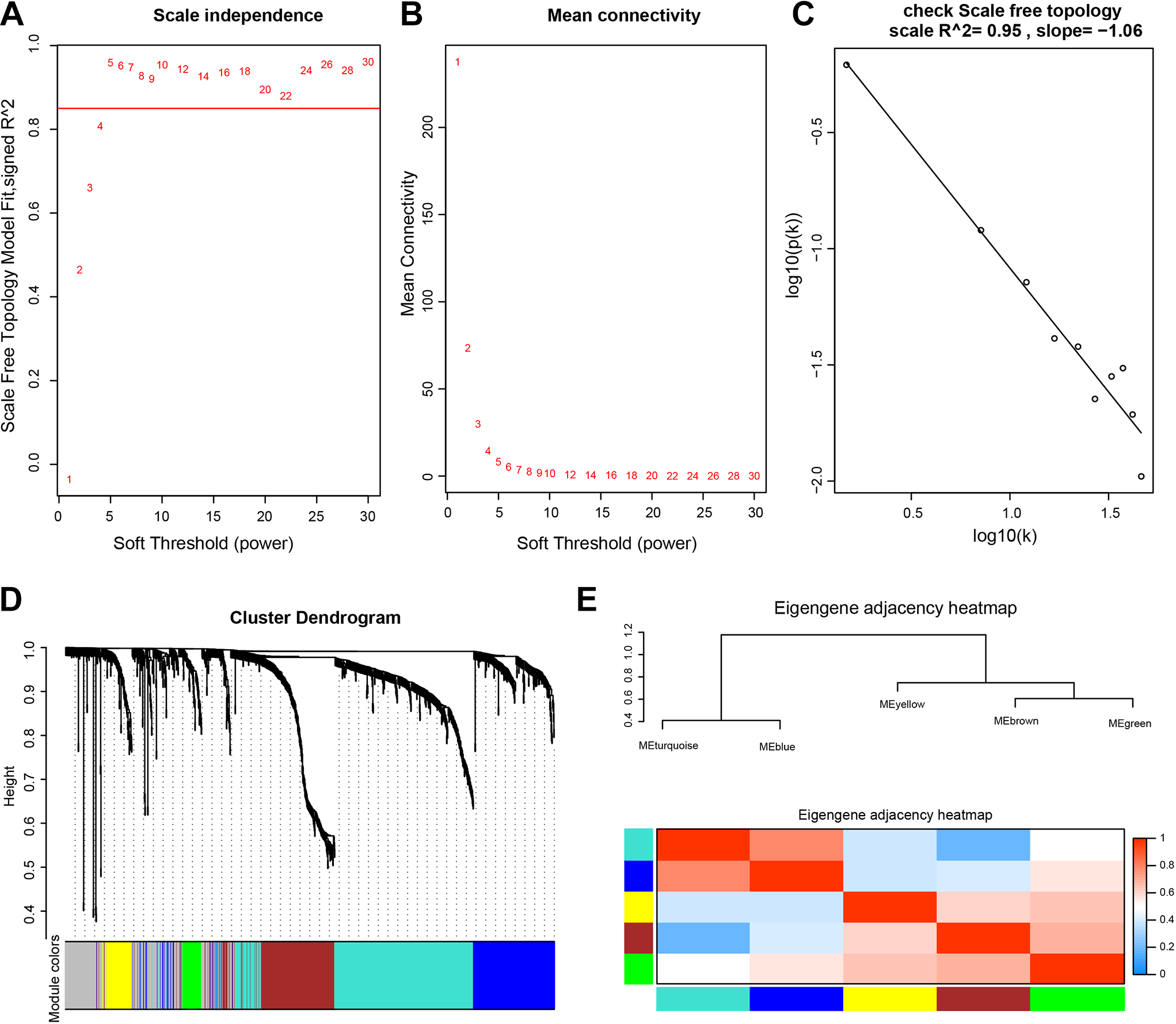
Analysis of the weighted gene correlation network analysis (WGCNA) of DEGs.A&B Analysis of network topology for various soft-threshold powers. The x-axis reflects the soft-thresholding power. The y-axis indicates the scale-free topology model fit index.
We mapped the relationships between the identified modules (Figure 3). The heatmap depicts the topological overlap matrix (TOM) among all genes included in the analysis. The darker red color represents a low overlap, and the progressively light color represents an increasing overlap. The results of this analysis indicated that the gene expression was relatively independent between modules.
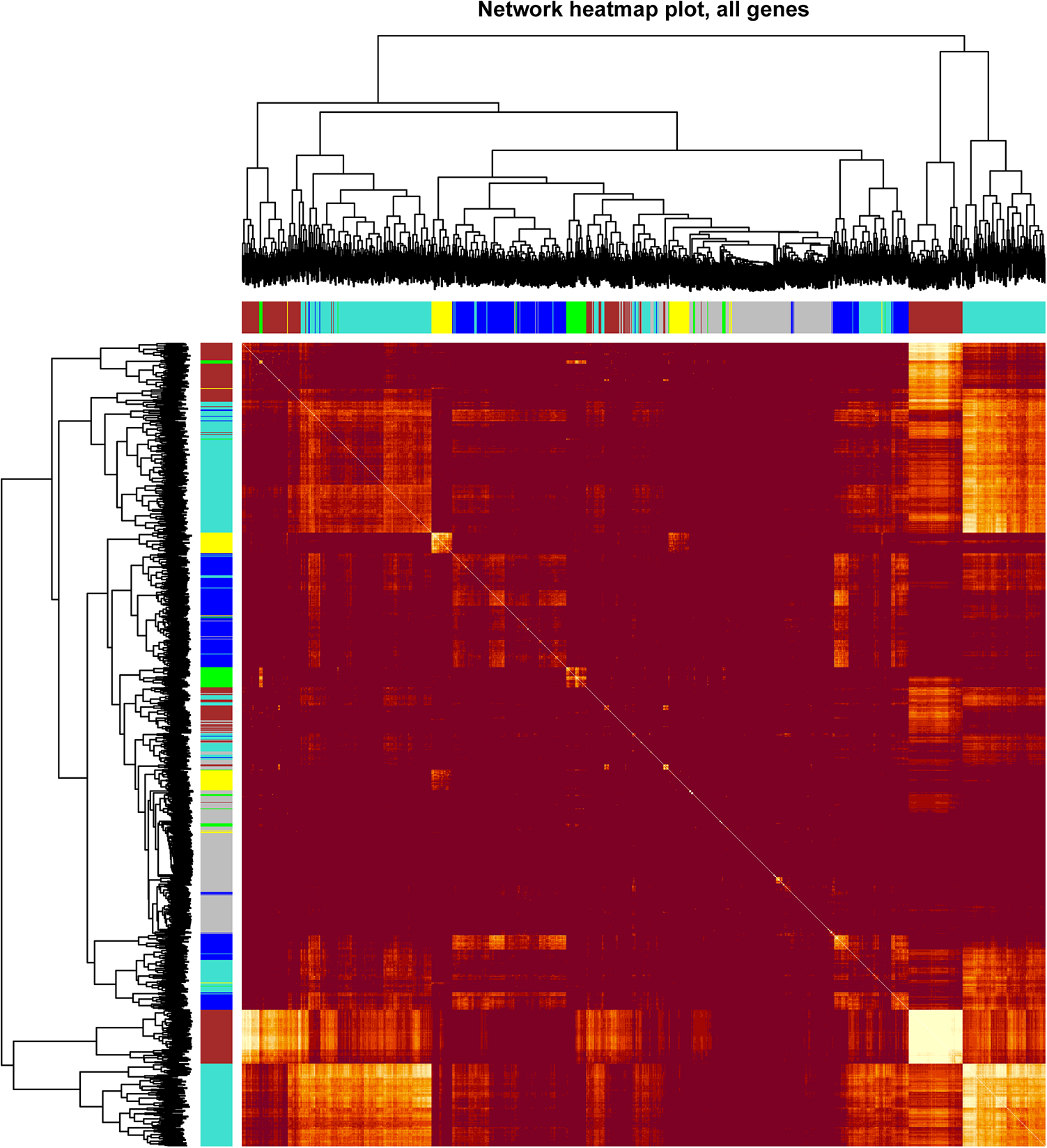
Visualization of the WGCNA network. Heatmap plot representing the gene network. The heatmap depicts the topological overlap matrix among all genes in the analysis.
Identification of Clinically Related Modules
We correlated modules with clinical characteristics (Figure 4A) and searched for the most significant associations. This analysis showed that the blue module was most significantly correlated with estrogen receptor status (Figure 4B). With the cor. gene GS > 0.2 and cor. Gene MM > 0.7 threshold limits, 51 out of 238 hub genes were identified (Figure 4C).

Module-trait associations. A, The trait heatmap of 838 BRCA patients. B, Module-trait relationships. Each row corresponds to a module, and each column corresponds to a trait. Each cell contains the corresponding correlation and P-value. C, Dot plot representing the genes’ gene significance and module membership in estrogen receptor status associated blue module.
The enrichment analysis was executed to describe the function of the critical genes (Supplemental Figure 1). The results indicated that the essential genes were significantly enriched in Hallmark KRAS signaling and IL-17 signaling pathway. These genes also enriched in several tumor-related terms, such as epithelial cell proliferation, germ cell development, and protein kinase activity, etc. It suggests the potential regulatory mechanism of these critical genes in BRCA.
Survival Analysis of Crucial Genes
To determine the prognostic performance of these genes, all of the 51 essential genes were tested by Kaplan-Meier analysis. It was found that 3 out of the 51 essential genes (FABP7, CXCL3, and LOC284578) were significantly associated with overall survival (OS) (Figure 5A). And 3 essential genes (FABP7, CAPN6, and NRG2) were significantly related to Relapse-free survival (RFS), too (Figure 5B). In particular, key gene FABP7 has dramatically associated with both OS and RFS. Finally, we obtained 5 essential prognostic associated genes (FABP7, CXCL3, LOC284578, CAPN6, and NRG2), which could be used as prognostic factors for BRCA. To further determine the prognostic efficacy of the 5 crucial genes, we used the OSbrca Tool to testify their survival ability. In overall survival group, we found FABP7 and CXCL3 were also associated with overall survival (OS) in GSE18229 and GSE39004. LOC284578 were not detected in these data sets (Supplemental Figure 2). In addition, FABP7, CAPN6, and NRG2 were related to Relapse-free survival (RFS) in the independent test data sets (GSE18229, GSE10893, GSE21653, and GSE2607) (Supplemental Figure 3).

Survival analysis of estrogen receptor status associated with crucial genes.
We also used GEPIA database 23 to testify the expression level of the 5 genes in BRCA tumor patients compare with normal samples (Supplemental Figure 4). We can see that all of the 5 genes were down-regulated in tumor, and the low expression of them in BRCA was related to the poor prognosis. Then, we used the HPA (The Human Protein Atlas) database to explore the expression of these genes in clinical patients. 24 Only FABP7 and NRG2 were found expressed in the BRCA tumor tissues by immunohistochemistry (IHC) analysis (Supplemental Figure 5). This suggests that the further study of us is using IHC to verify the expression of these genes in clinical patients. All these results showed that the 5 genes played a crucial role in BRCA prognosis.
Discussion
Invasive breast cancer is the most common malignancy in females and seriously threatens physical and mental health. 25 -27 In recent years, bioinformatics analysis has been widely used in cancer research. 28 -30 This kind of research using a public database containing many samples provides an excellent way to identify tumor biomarkers. 31 -33
In this study, we built the co-expression modules via WGCNA using the TCGA BRCA data. We identified genes that were significantly differentially expressed in BRCA patients compared with normal samples. Then the function enrichment was investigated to show these genes’ functions. After WGCNA analysis, we obtained a gene module that associated with estrogen receptor status, and several hub genes in the module were survival associated. FABP7 has been reported to be a key metabolic regulator in HER2+ breast cancer brain metastasis 34 and could mediate triple-negative breast cancer cell death via PPAR-α signaling. 35 CXCL3 was found to be a potential target for breast cancer metastasis 36 and inhibition of CXCL3 could reduce STAT3 activation. 37 LOC284578 has not been reported to be related to the prognosis of breast cancer in previous studies, which may be a new prognostic biomarker of BRCA. CAPN6 could regulate RAC1 activity and cell motility through interaction with GEF-H1. 38 It also can be regulated by PI3K-Akt pathway and may be a therapeutic target of cancer. 39 TGFα, HB-EGF, and NRG2 have been reported to be related to the biological aggressiveness of the tumors. 40 The genetic variants in NRG2 also can influence breast cancer risk. 41 In summary, the 5 genes identified in our study may use as new biomarkers for improving the prognosis of BRCA patients.
Our research does have some limitations. First of all, our research results are based on public online database information, so we need to prove it through experiments. In the future, we will build a cell model to express the above 5 genes to verify our conclusions differentially. Second, although we conducted gene enrichment analysis, our research did not clarify the mechanism of these critical genes participating in BRCA. Therefore, this is one of the focuses of our future examinations.
Supplemental Material
Supplemental Material, Supplemental_Figures - Transcriptomics and Prognosis Analysis to Identify Critical Biomarkers in Invasive Breast Carcinoma
Supplemental Material, Supplemental_Figures for Transcriptomics and Prognosis Analysis to Identify Critical Biomarkers in Invasive Breast Carcinoma by Jun Wu, Xiao-Jun Liu, Jia-Nan Hu, Xu-Hui Liao and Fei-Fei Lin in Technology in Cancer Research & Treatment
Footnotes
Authors’ Note
Jun Wu and Xiao-Jun Liu contributed equally to this work. The study was based on an analysis of public data and did not address any ethical issues.
Acknowledgments
The authors wish to thank the 4 anonymous reviewers whose constructive comments were helpful to strengthen the presentation of this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.