
Abstract
BACKGROUND:
Persistent infection of high-risk HPVs is known to cause diverse carcinomas, mainly cervical, oropharyngeal, penile, etc. However, efficient treatment is still lacking.
OBJECTIVE:
Identify and analyze potential therapeutic targets involved in HPV oncogenesis and repurposing drug candidates.
METHODS:
Integrative analyses were performed on the compendium of 1887 HPV infection-associated or integration-driven disrupted genes cataloged from the Open Targets Platform and HPVbase resource. Potential target genes are prioritized using STRING, Cytoscape, cytoHubba, and MCODE. Gene ontology and KEGG pathway enrichment analysis are performed. Further, TCGA cancer genomic data of CESC and HNSCC is analyzed. Moreover, regulatory networks are also deduced by employing NetworkAnalyst.
RESULTS:
We have implemented a unique approach for identifying and prioritizing druggable targets and repurposing drug candidates against HPV oncogenesis. Overall, hundred key genes with 44 core targets were prioritized with transcription factors (TFs) and microRNAs (miRNAs) regulators pertinent to HPV pathogenesis. Genomic alteration profiling further substantiated our findings. Among identified druggable targets, TP53, NOTCH1, PIK3CA, EP300, CREBBP, EGFR, ERBB2, PTEN, and FN1 are frequently mutated in CESC and HNSCC. Furthermore, PIK3CA, CCND1, RFC4, KAT5, MYC, PTK2, EGFR, and ERBB2 show significant copy number gain, and FN1, CHEK1, CUL1, EZH2, NRAS, and H2AFX was marked for the substantial copy number loss in both carcinomas. Likewise, under-explored relevant regulators, i.e., TFs (HINFP, ARID3A, NFATC2, NKX3-2, EN1) and miRNAs (has-mir-98-5p, has-mir-24-3p, has-mir-192-5p, has-mir-519d-3p) is also identified.
CONCLUSIONS:
We have identified potential therapeutic targets, transcriptional and post-transcriptional regulators to explicate HPV pathogenesis as well as potential repurposing drug candidates. This study would aid in biomarker and drug discovery against HPV-mediated carcinoma.
Introduction
Human papillomaviruses (HPVs) are the small circular double-stranded DNA (dsDNA) onco-virus from the Papillomaviridae family. Based on the malignant risks, these are further divided into two subgroups, i.e., high-risk HPVs (HR-HPVs) that are highly carcinogenic in nature and low-risk HPVs (LR-HPVs) mainly associated with benign warts and lesions [1, 2]. Persistency of HR-HPVs infection is highly crucial in the cancer progression towards precancer and invasion [2, 3]. HPVs are reported to cause distinct cancers such as cervical, oropharyngeal, penile, vulvar, vaginal, and anal carcinomas, etc. [1, 4]. Two HPV oncogenes, i.e., E6 and E7, are mainly involved in HPV oncogenesis [5]. HR-HPV types 16 and 18 play a vital role in the progression and etiology of cervical cancer and head and neck squamous cell carcinomas (HNSCCs) [1, 5].
Cervical cancer is the fourth most common cancer in women (
Along with the HPV infection and multi-stage progression of HPV-attributable cancers, diverse events and consequences are interconnected. This mainly includes HPV integration events, deregulation of miRNAs, epigenetic modifications, and genomic alterations [8, 12, 13]. One of the critical events is HPV integration, which contributes to carcinogenesis. Integration event leads to the disruption and alterations of host genes and enhances genomic instability [8, 12, 14].
Various studies reported the distribution and role of HPVs in different carcinomas [1, 13, 15, 16, 17]. Simultaneously, high-throughput data from the multiple cohorts of distinct carcinomas were analyzed for the genomic, transcriptomic, and epigenomic events [13, 15, 16, 18, 19]. The landscape of genomic alterations was analyzed in cervical carcinoma [15]. The Cancer Genome Atlas (TCGA) Research Network provides integrated genomic and molecular characterization of 228 cervical cancer samples [20]. Stransky et al. reveal a mutational spectrum among genes in HNSCC utilizing large-scale sequencing [16].
Furthermore, several studies also describe data analysis mainly through differential expressed genes (DEGs) identification to elucidate potential targets in different cancers, including cervical and HNSCC, utilizing different bioinformatics approaches [21, 22, 23, 24]. This assimilates distinct strategies like differentially expressed genes (RNAseq and microarray), protein-protein interactome, co-expression networks, GO and pathway enrichment, protein expression, genomic (mutational profile, copy number variations (CNVs), mRNA regulation), epigenomic alterations, etc. [21, 23, 24, 25, 26, 27, 28, 29].
Moreover, different efforts are also made to prevent and treat HPV-associated diseases through developing vaccines and drugs against HPVs and cancers [30, 31]. Currently, there are three prophylactic vaccines have been approved to counter HPV infections. These three recombinant vaccines are as follows, a bivalent HPV-16/18 vaccine named GlaxoSmithKline’s Cervarix
In this study, an integrative approach is employed to elucidate HPV oncogenesis with a focus on TCGA genomic data from cervical squamous cell carcinoma (CSCC) and head and neck squamous cell carcinoma (HNSCC). Known HPV infection-associated candidate genes were analyzed [36]. Notably, genes disrupted due to integration events in HPV pathogenesis [12] are also included in the meta-analysis. Our findings related to potential therapeutic targets, hallmark molecular functions, enriched pathways, genomic alterations, transcription factors, miRNAs, and potential repurposing drugs could assist biomarker discovery and therapeutic development for HPV-linked carcinomas.
Analyses design and workflow of the study.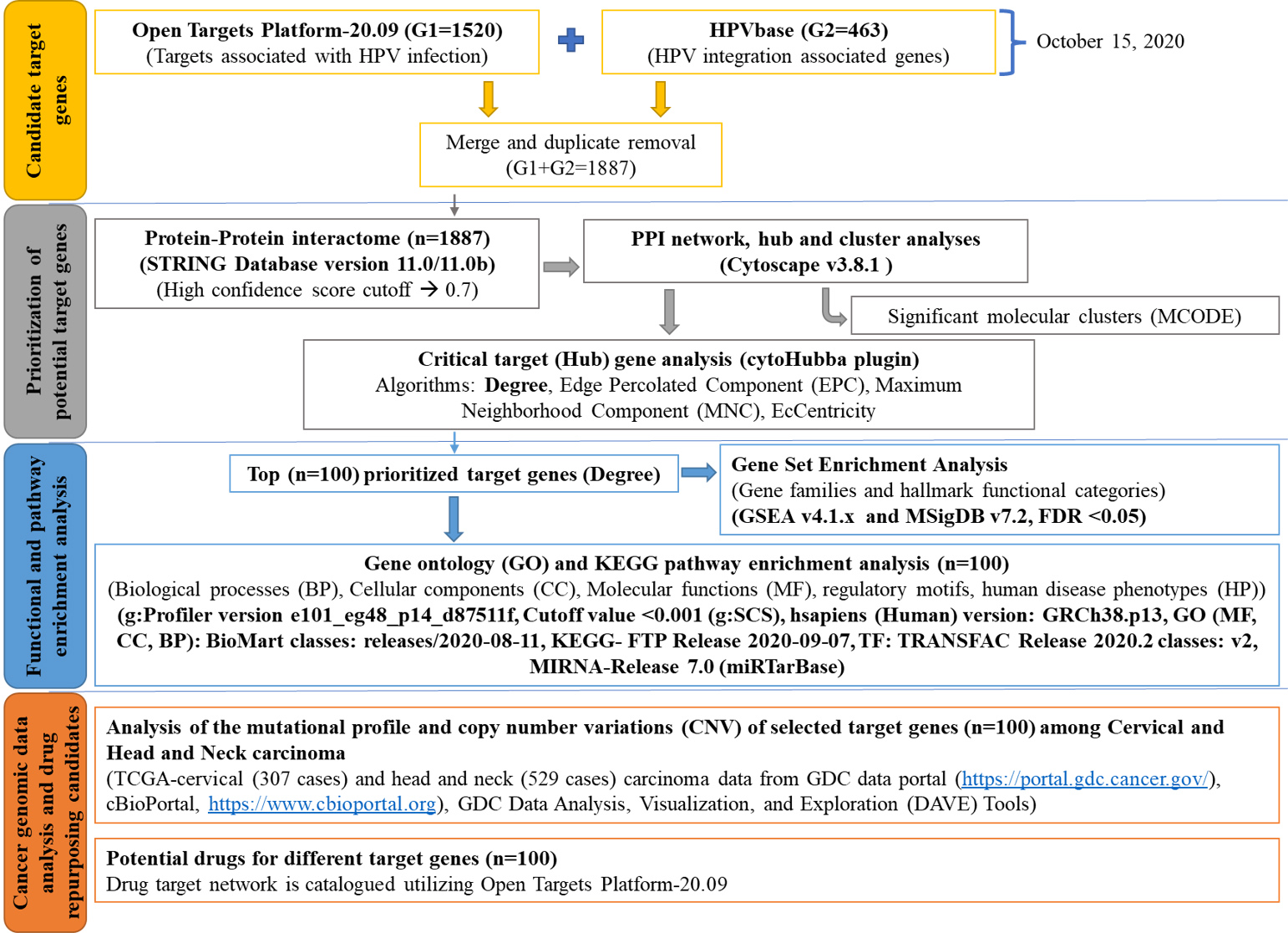
The complete study design and workflow are illustrated in Fig. 1. All the steps of the workflow are further elaborated below. The relevant study data is available through the GitHub repository (
Selection of candidate target genes
We have retrieved all the candidates from the two sources. First, we have extracted the genes from the “Open Targets Platform v20.09,” which provides disease-specific targets [36]. Overall, 1520 (G1 list) targets associated with the HPV infection were obtained. Second, a set of genes disrupted due to the HPV integration events were acquired from our previous resource, “HPVbase” [12]. Overall, 463 (G2 list) HPV-integration associated genes were obtained. We have combined the G1 and G2 lists to catalog the final working set. Ninety-six duplicates were found between G1 and G2 and removed. In total, 1887 candidate genes were utilized for further downstream analysis (Table S1).
Protein-protein interaction (PPI) network-based prioritization of potential target genes
All the 1887 candidate genes were searched by implementing the Search Tool for the Retrieval of Interacting Genes (STRING) database v11 [37] to identify protein-protein interactome with a high confidence score cutoff of 0.7. Further, the obtained interacting proteins were analyzed utilizing the Cytoscape program v3.8.1 [38] and different modules, mainly cytoHubba [39] and MCODE [40]. From the PPI network, significant molecular clusters were identified using MCODE, and a minimum degree cutoff of 10 was chosen. Furthermore, to determine the critical target (Hub) genes from the OncoHPV-PPI network, cytoHubba analysis utilizing the four different algorithms, i.e., Degree, Edge Percolated Component (EPC), Maximum Neighbourhood Component (MNC), and EcCentricity is performed.
Gene ontology (GO) and pathways
Further, GO, and Kyoto Encyclopedia of Genes and Genomes (KEGG) [41] pathway enrichment analysis was performed on the prioritized target genes to explore the systematic functional and genomic annotations and pathway information. This includes identifying the critical biological processes, cellular components, molecular functions, and significant pathways. For this, g:profiler version e101_eg48_p14_d87511f [42] was employed for the systematic analyses, which provide multiple data sources for the comprehensive illustrations. A stringent cutoff value
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis was conducted to detect different gene families and define top functional categories through hallmarks gene sets from Molecular Signatures Database (MSigDB) v7.2 [43] with a significant cutoff value, i.e., FDR
Analysis of the mutational profile and copy number variations (CNVs) of selected target genes among cervical and head and neck carcinoma
Further, genomic alterations (mutations and CNVs) among the key target genes were explored on the TCGA-cervical and head and neck carcinoma. Data from the GDC data portal (
Potential drugs for different target genes
Target genes were also analyzed for the potential drugs targeting these proteins utilizing the Open target platform v20.09 [36]. The Drug-target network is cataloged and presented. Simultaneously, protein-protein relationships were also explored using the OmniPath DB combining the 115 databases [48].
Transcriptional and post-transcriptional regulatory analyses
We have further analyzed the gene regulatory network utilizing NetworkAnalyst 3.0 [49]. For this, gene-miRNAs, gene-TFs, and miRNAs-gene-TFs interactions were identified employing miRTarBase v8.0 [50], JASPAR [51], and RegNetwork [52] resources, respectively. A regulatory relationship network was constructed using NetworkAnalyst. In miRNAs-gene-TFs co-regulatory interactions, a minimum network option is utilized for simplifying the dense network.
Results
OncoHPV-PPI Network and prioritization of target genes
In total, 1520 HPV infection-associated genes were obtained from the “Open Targets Platform” and 463 HPV integration disrupted genes from “HPVbase.” After removing 96 duplicates from the list, 1887 candidate genes were utilized for the PPI network analysis using the String database and Cytoscape. With the (high) confidence score cutoff of 0.7, the final PPI network had 1879 nodes and 20735 edges. Different nodes represent proteins in the network, and edges show associations among proteins. The average node degree is 22.1, and the PPI enrichment
All the connected nodes in the PPI Network were analyzed with the CytoHubba application to explore the hub (target) genes. Top 100 genes from four different algorithms, i.e., Degree (Fig. S1), Edge Percolated Component (EPC), Maximum Neighbourhood Component (MNC), and EcCentricity, were retrieved (Table S2). Genes from each algorithm are integrated to deduce the 44 core genes (Fig. S2 and Table 1). Further, taking degree as the significant criteria, the top 100 genes from the PPI network (Fig. S1) were utilized for the different downstream analyses.
Table representing the integration of significant targets from four different algorithms
Table representing the integration of significant targets from four different algorithms
Abbreviations: EPC, edge percolated component; MNC, maximum neighbourhood component.
Identified top 100 target genes were subjected to the Gene Set Enrichment Analysis (GSEA) investigated through the Molecular Signatures Database (MSigDB) [43]. Gene families and functional categories of these genes are cataloged. Overall, 70 genes were categorized in the seven gene families, i.e., Oncogenes (21 genes), Transcription factors (TFs) (20), Protein kinases (19), Cytokines and growth factors (15), Translocated cancer genes (11), Tumor suppressors (8), and Cell differentiation (CD) markers (5). Target genes and corresponding gene families were shown using the Sankey plot (Fig. 2).
Different functional categories and gene families among target genes (Sankey plot).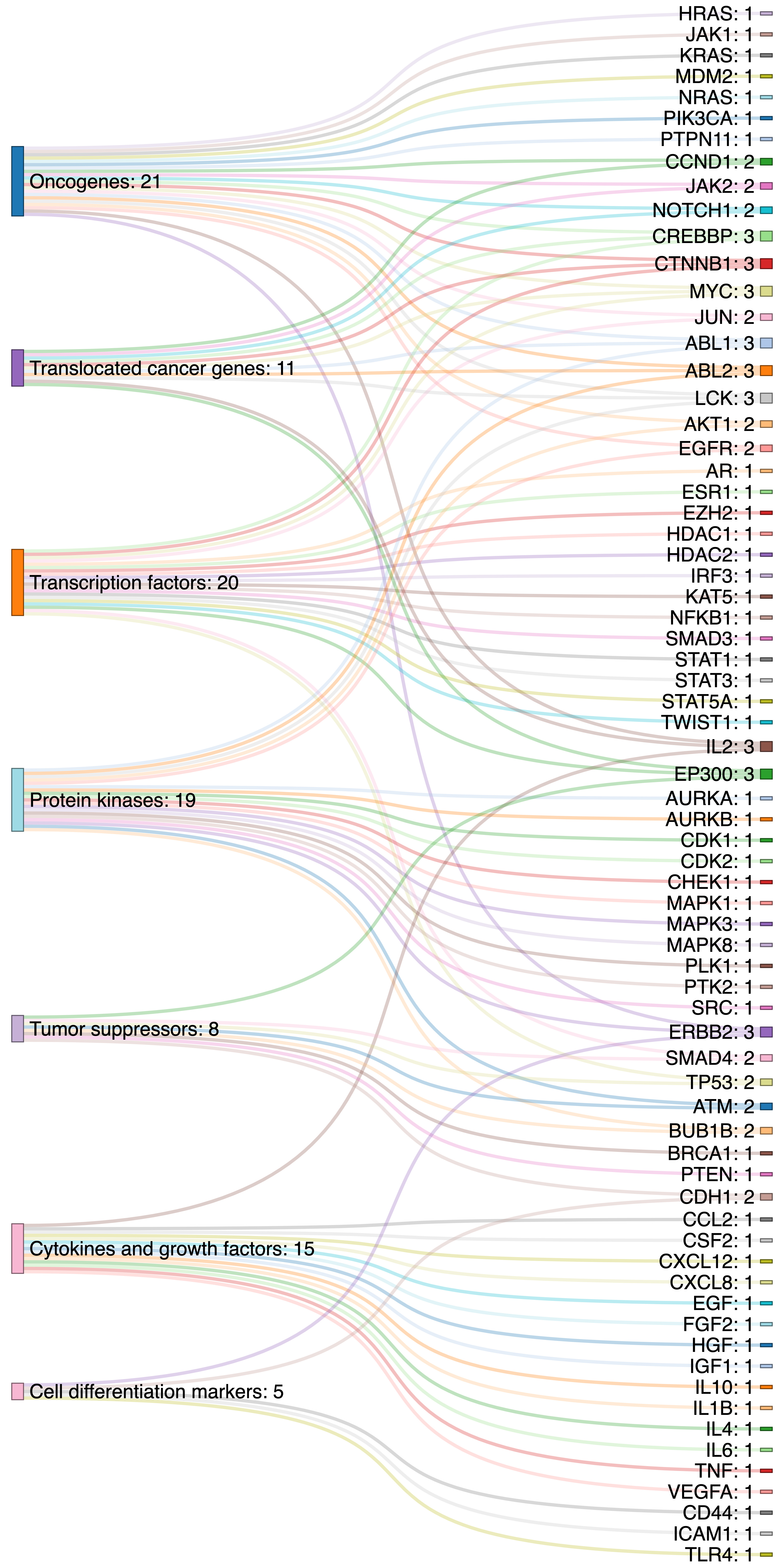
Enrichment map of GO: MF, GO: BP, GO: CC, KEGG pathways, miRNAs, and the human phenotype ontology. The most significant features in each category are marked and listed in the figure.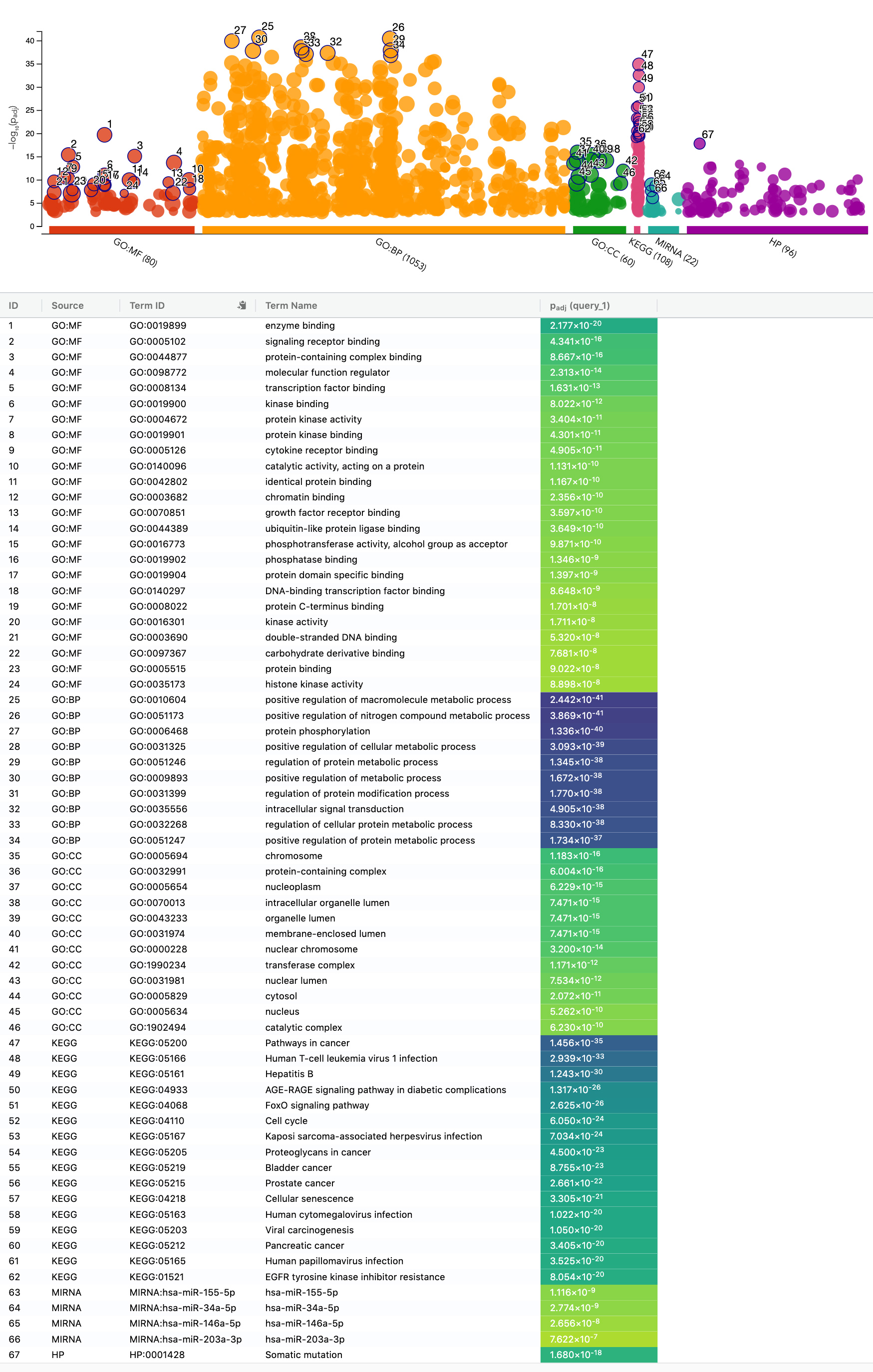
Simultaneously, the top 20 enriched potential functional sets were identified (Table S3). The most significant three hallmarks functional sets among the target genes are G2M-Checkpoint (FDR q-value: 6.58E-25), E2F-Targets (3.60E-20), and apoptosis (3.60E-20). G2M-Checkpoint includes 20 genes (MYC, CDK1, CDC20, MAD2L1, RPA2, AURKA, CCNB2, EZH2, H2AX, PLK1, CHEK1, AURKB, CCND1, SMAD3, CUL1, CCNA2, CDC6, KIF11, EGF, UBE2C) from targets. E2F-Targets include 17 genes, i.e., MYC, CDK1, CDC20, MAD2L1, RPA2, AURKA, CCNB2, EZH2, H2AX, PLK1, CHEK1, AURKB, BRCA1, CDKN1A, TP53, PCNA, and BUB1B. Likewise, 16 genes, i.e., CCND1, BRCA1, CDKN1A, IL6, IL1B, TNF, CD44, JUN, CDK2, CASP3, CTNNB1, PTK2, APP, ERBB2, CREBBP, and HGF are marked under the apoptosis hallmark. Other important hallmarks include signaling (TNFA_SIGNALING_VIA_NFKB, PI3K_AKT_MTOR, IL6_JAK_STAT3, WNT_BETA_CATENIN, and NOTCH SIGNALING), MYC_TARGETS_V1, and P53_PATHWAY (Table S3).
Enrichment analysis of the top 100 genes (degree) is performed. g-profiler is employed to search significant molecular functions (MF), biological process (BP), cellular components (CC), KEGG biological pathways, regulatory motifs (miRTarBase), and human disease phenotypes (HP) in humans utilizing recommended “g:SCS” method with the stringent threshold of 0.001 (Fig. 3). Overall, 80 molecular functions were determined, and the most significant GO: molecular function is enzyme binding (GO:0019899) with adjusted
In total, 1053 biological processes were deduced, and the most significant GO: biological process is positive regulation of macromolecule metabolic process (GO:0010604), having 2.44E-41 adjusted
OncoGrid presents the top 50 mutated target genes (Y-axis) and 200 most mutated cases (X-axis) with genomic alterations (mutations and CNVs) frequencies in CESC along with clinical information and data availability (bottom). Gene-wise mutation consequences and CNVs are shown horizontally, and case-wise, are shown vertically in each cell of the grid. Mutations color code: missense (aqua), frameshift (maroon), start lost (amber), stop gained (purple), and stop lost (grey). CNVs: loss (blue) and gain (red).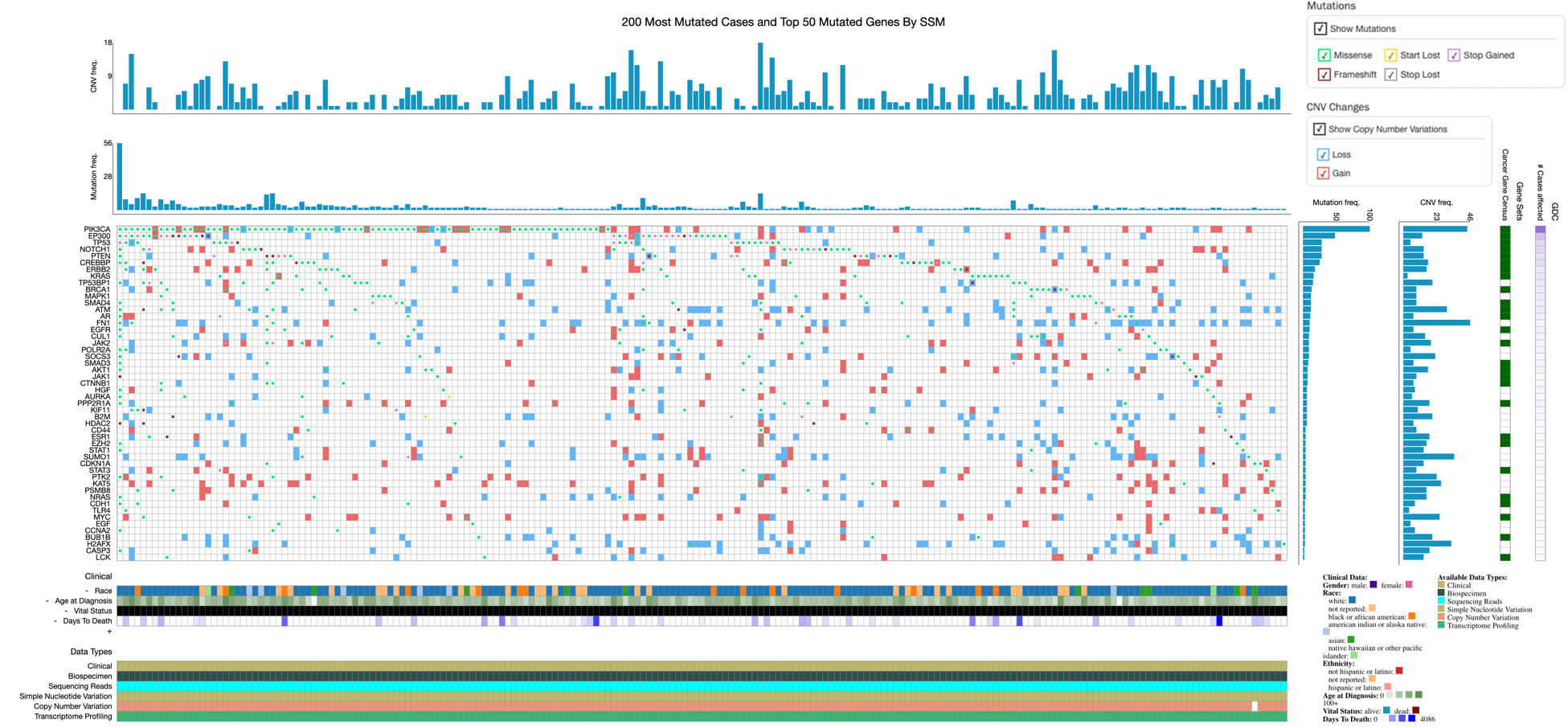
OncoGrid depicting the top 50 mutated target genes (Y-axis) and 200 most mutated cases (X-axis) with genomic alterations (mutations and CNVs) frequencies in HNSCC along with clinical information and data availability (bottom). Gene-wise mutation consequences and CNVs are shown horizontally, and case-wise, are shown vertically in each cell of the grid. Mutations color codes: missense (aqua), frameshift (maroon), start lost (amber), stop gained (purple), and stop lost (grey). CNVs: loss (blue) and gain (red).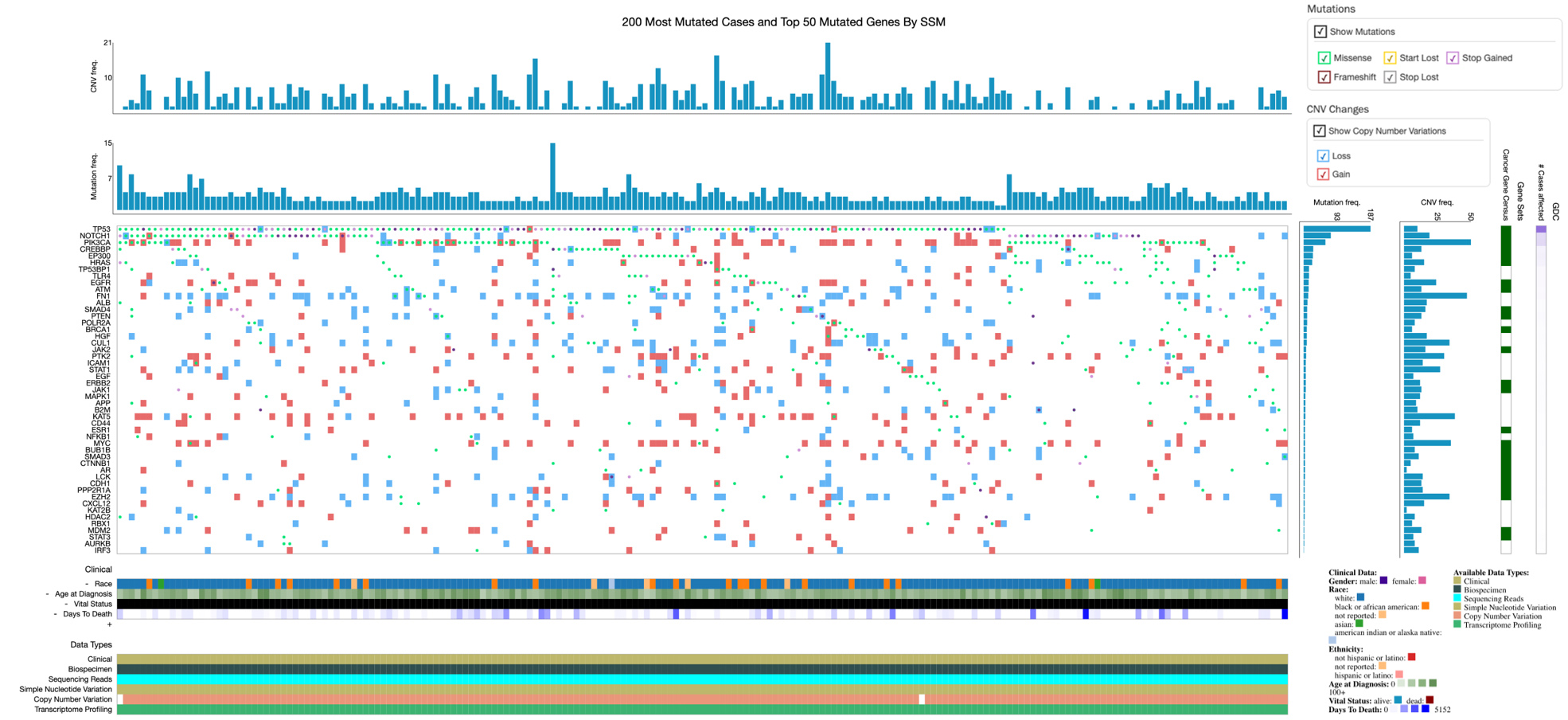
FGF2, CD44, ICAM1, SOCS3, ESR1, HDAC2, IL2, UBE2C).
Further, 108 biological pathways were identified, and the most significant KEGG biological pathway was Pathways in cancer (KEGG:05200) with adjusted
Likewise, 22 regulatory miRNAs were marked, and the most significant is hsa-miR-155-5p (adjusted
The Cancer Genome Atlas (TCGA)-PanCancer Atlas (PCA) (
Out of 307 CESC cases, 293 samples show mutation (280 cases) or CNV data (284 cases) for the target genes. OncoGrid of genomic alterations in CESC provides a comprehensive view of 50 frequently mutated genes with CNVs (loss and gain) among samples (Fig. 4). The top 20 most frequently mutated genes are presented in Fig. S3. The most frequently mutated genes in the CESC among selected targets are PIK3CA (3q26.32), EP300 (22q13.2), PTEN (10q23.31), CREBBP (16p13.3), NOTCH1 (9q34.3), TP53 (17p13.1), KRAS (12p12.1), ERBB2 (17q12), TP53BP1 (15q15.3), SMAD4 (18q21.2), AR (Xq12), MAPK1 (22q11.22), POLR2A (17p13.1), EGFR (7p11.2), BRCA1 (17q21.31), ATM (11q22.3), FN1 (2q35), ESR1 (6q25.1, 6q25.2), CUL1 (7q36.1), and PPP2R1A (19q13.41) (Table S5). In total, 860 mutations on target genes were marked. Most mutations are present on EP300, PTEN, NOTCH1, CREBBP, PIK3CA, and TP53 (Table S5). The top 20 genes with the highest number of mutations are depicted in Fig. S4. Likewise, genes with high CNVs (gain and loss) are shown in Figs S5 and S6.
Drug-target network illustrating available drug repurposing candidate molecules (150) against 17 key target genes.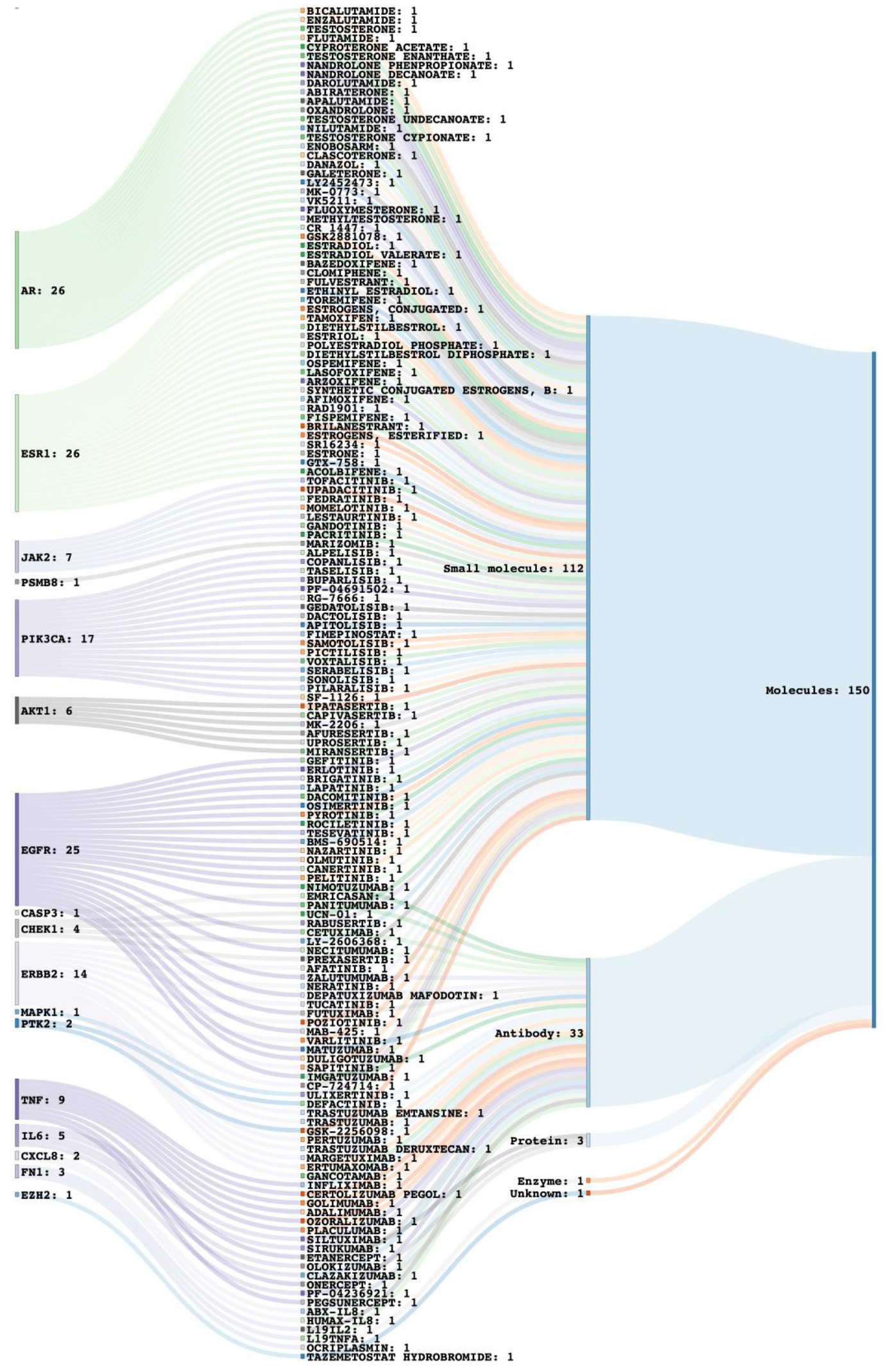
Network illustrating Gene-TFs regulatory interactions having 183 nodes. Red depicts target genes (99), and cyan depicts TFs (84).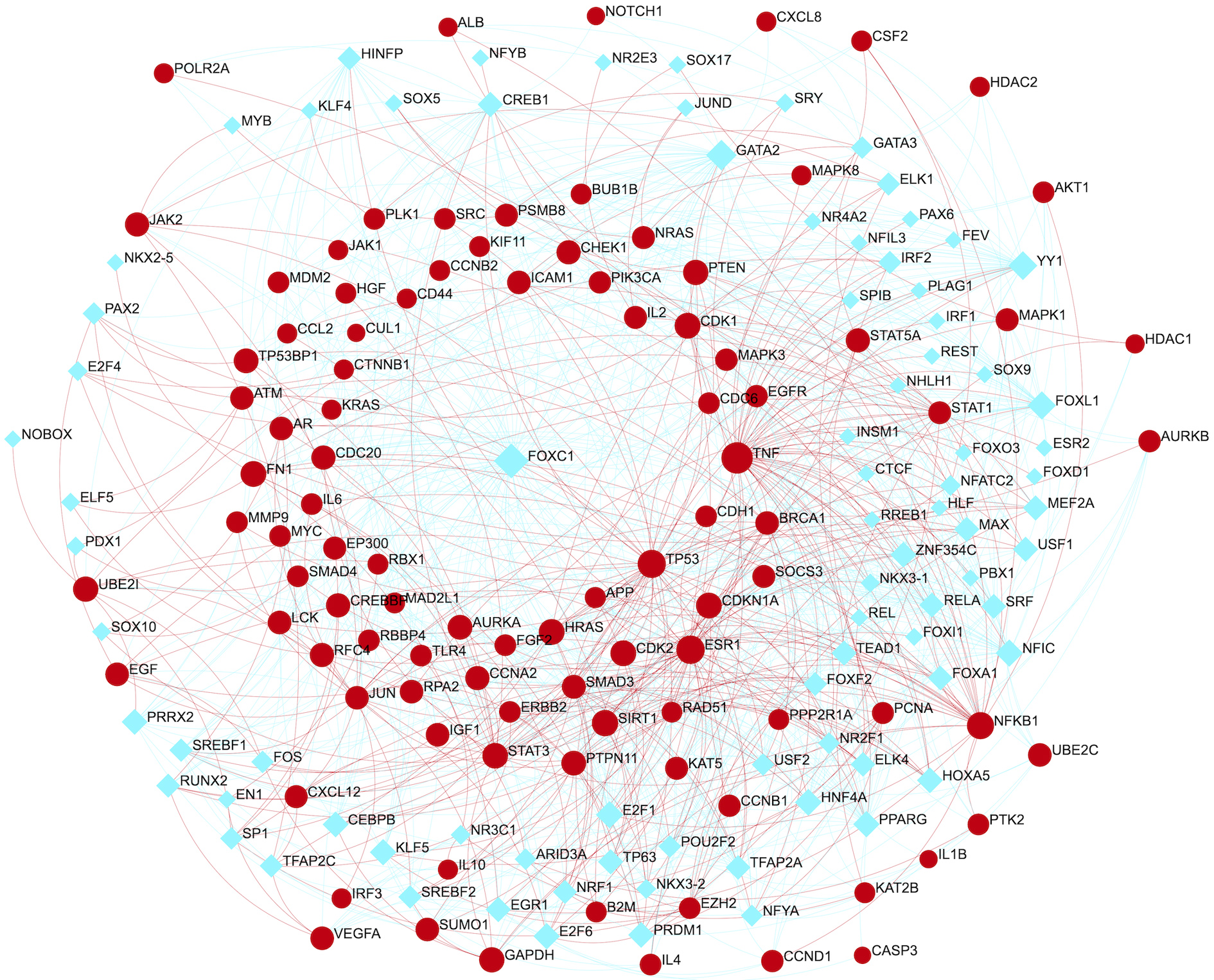
Interaction network depicting significant Target-miRNAs regulatory associations. Targets shown in blue lined circles and square blocks (cyan) depict miRNAs.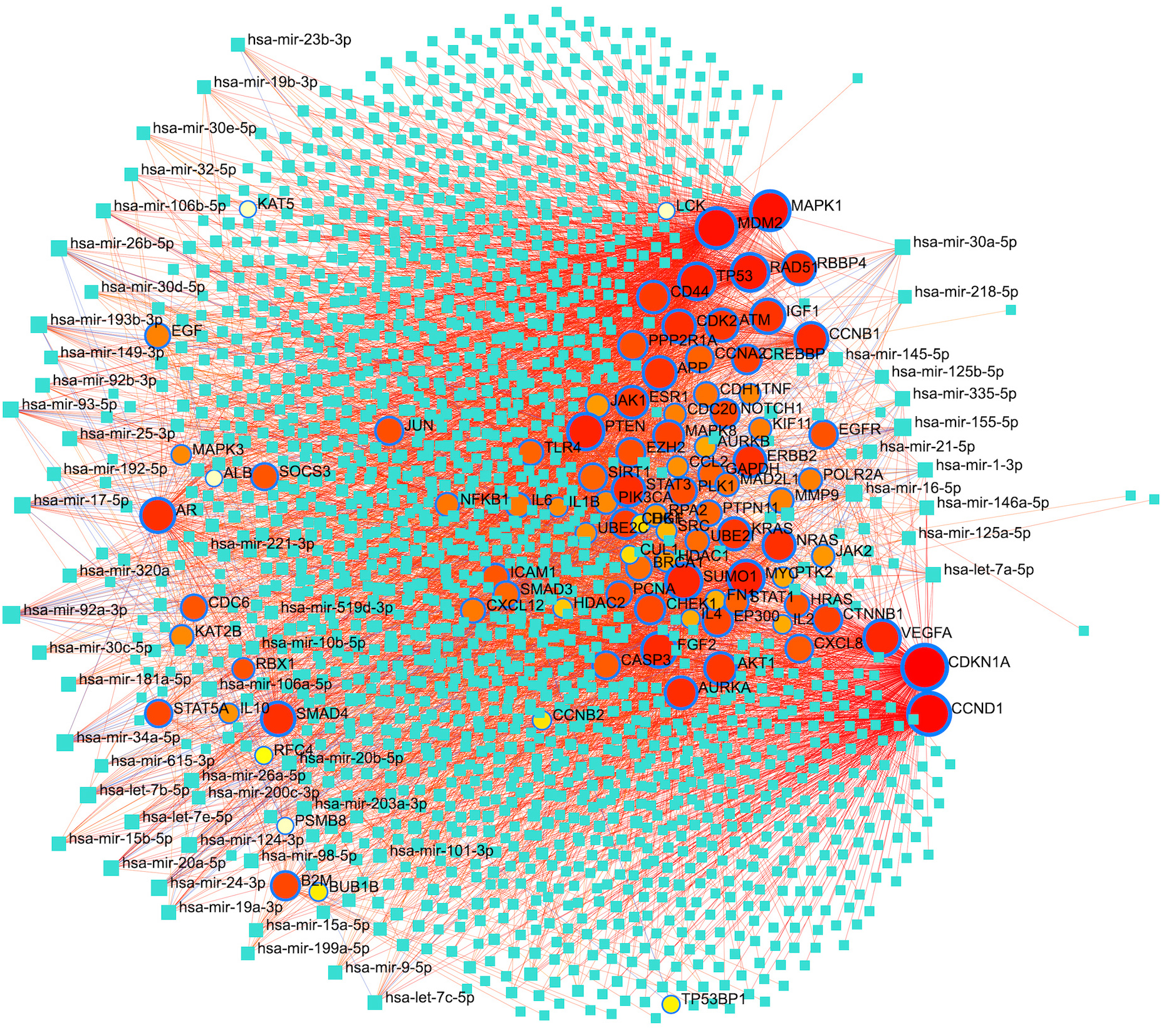
Network showing significantly co-regulated elements, i.e., miRNAs, targets, and transcription factors. Blue indicates miRNAs (left), orange shows targets (center), and the green depicts TFs (right).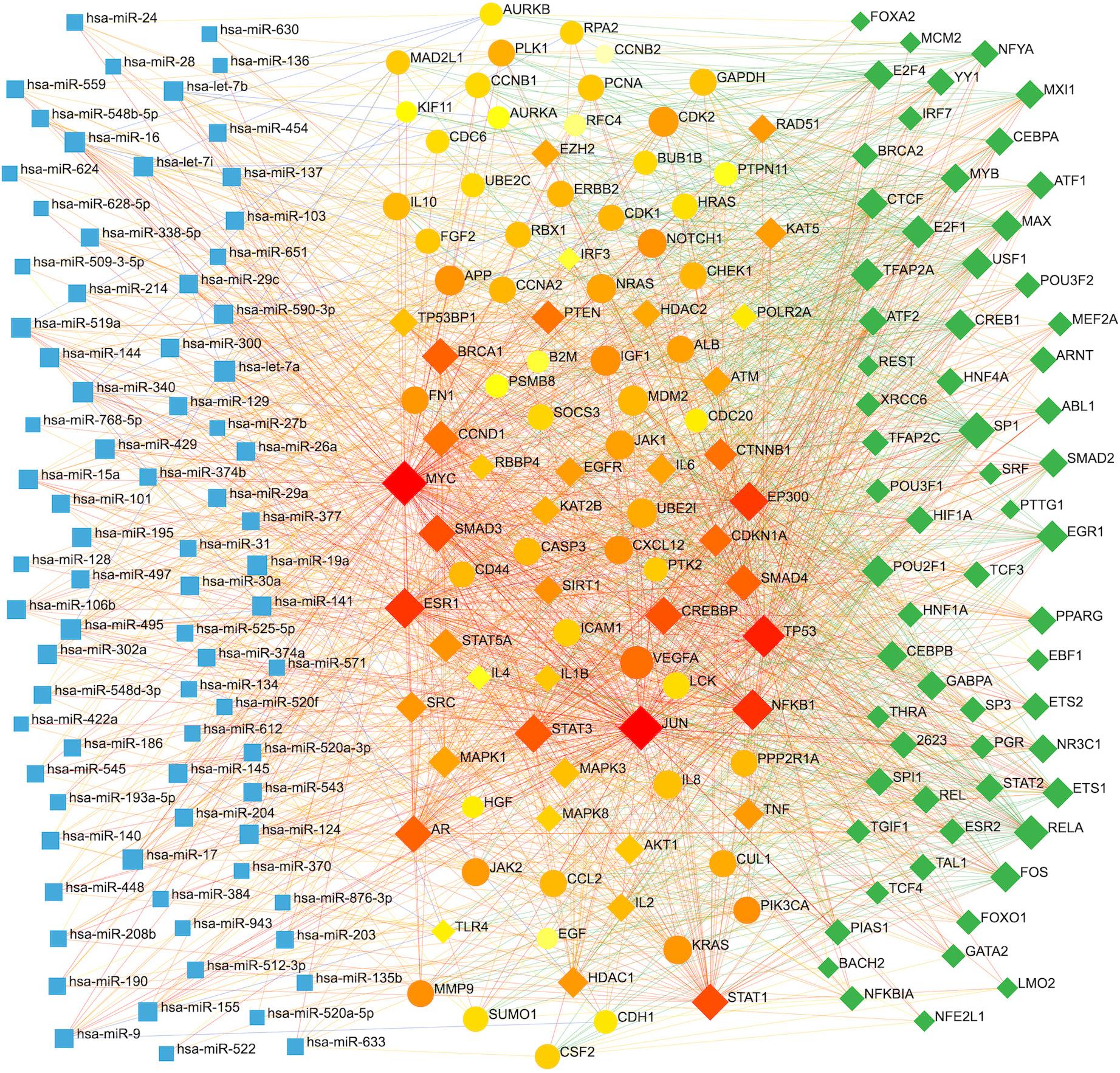
Likewise, from 529 HNSCC samples, 524 cases have mutations (503 cases) or CNV data (515 cases) for undertaken target genes. OncoGrid of these alterations from HNSCC samples represents a complete landscape of 50 frequently mutated genes along with CNVs (loss and gain) (Fig. 5). The top 20 most frequently mutated genes are illustrated in Fig. S7. Frequently mutated genes among all are TP53 (17p13.1), NOTCH1 (9q34.3), PIK3CA (3q26.32), EP300 (22q13.2), CREBBP (16p13.3), HRAS (11p15.5), TLR4 (9q33.1), EGFR (7p11.2), FN1 (2q35), ATM (11q22.3), TP53BP1 (15q15.3), PTEN (10q23.31), POLR2A (17p13.1), SMAD4 (18q21.2), BRCA1 (17q21.31), EGF (4q25), HGF (7q21.11), ERBB2 (17q12), STAT1 (2q32.2), and PTK2 (8q24.3) (Table S6). Overall, 1092 mutations are present in target genes. The most mutated genes are TP53, NOTCH1, CREBBP, PIK3CA, EP300, TLR4, EGFR, FN1, TP53BP1, and ATM (Table S6). The number of mutations in most mutated genes is presented in Fig. S8. Similarly, Figs S9 and S10 depict high CNV (gain and loss) genes, respectively.
We have also analyzed the cross-mutated target genes between CESC and HNSCC samples. Overall, 49 common mutations were found between both the carcinomas (CESC (103 samples), HNSCC (130) of 20 genes (Fig. S11 and Table S7). The most frequently mutated genes are PIK3CA, TP53, MAPK1, EP300, PTEN, and ERBB2 (Fig. S11).
The key target genes were analyzed for the potential drugs targeting these proteins utilizing the Open target platform. The drug-target relationship is cataloged (Table S8). Overall, we identified 230 drugs pertaining to 41 target proteins among 100. These drugs belong to different molecule types, i.e., small molecules (161), Antibody (58), Protein (7), Enzyme (1), Oligosaccharide (1), and unknown (2) (Fig. S12A). Among all, maximum drugs are targeting AR (26), ESR1 (26), EGFR (25), PIK3CA (17), and ERBB2 (14) (Fig. S12B). The drug-target network for the 17 key targets with 150 available drug molecules is depicted in Fig. 6. This also illustrates that 66 drug molecules correspond to the seven most promising draggable targets. Additionally, interactions between targets were also obtained utilizing the OmniPath DB, which provides comprehensive knowledge from 115 databases focusing on signaling pathways. This provides 707 enzyme-substrate relationships, 150 pathway-protein interactions, and 355 protein-protein interactions between targets (Fig. S13).
Transcriptional and post-transcriptional regulations
Gene-TFs (Transcriptional), gene-miRNAs (post-transcriptional) and miRNAs-gene-TFs co-regulatory relationships were identified. In transcriptional regulation, we have identified 84 transcription factors pertaining to 99 targets. Gene-TFs regulatory network with 183 nodes is shown in Fig. 7. Among these, FOXC1, GATA2, YY1, FOXL1, E2F1, NFIC, PPARG, CREB1, SRF, RELA, TFAP2A, HINFP, SREBF1, POU2F2, USF2, and NFYA are most promising TFs associated to targets (Table S9).
Further, a target-miRNAs regulatory relationship is deduced, and a network is constructed. In total, 1757 regulatory miRNAs were identified pertaining to targets. Significant target-miRNAs interactions are shown in Fig. 8. The most engaging miRNAs are hsa-mir-92a-3p, hsa-mir-155-5p, hsa-mir-34a-5p, hsa-mir-16-5p, hsa-mir-193b-3p, hsa-let-7b-5p, hsa-mir-17-5p, hsa-mir-24-3p, hsa-mir-26b-5p, hsa-mir-93-5p, hsa-mir-124-3p, hsa-mir-20a-5p, hsa-mir-30a-5p, hsa-mir-106a-5p, hsa-let-7a-5p, hsa-mir-26a-5p, hsa-mir-335-5p, hsa-mir-1-3p etc. (Table S10).
Likewise, we have further explored miRNAs-target-TFs co-regulations. Overall, 78 and 64 significantly co-regulated miRNAs and TFs were identified as associated with targets, respectively (Table S11). miRNAs-target-TFs co-regulatory interactions are depicted in Fig. 9. The most co-regulated miRNAs are hsa-let-7a, hsa-miR-495, hsa-miR-340, hsa-miR-16, hsa-miR-17, etc. Likewise, SP1, RELA, TFAP2A, E2F1, POU2F1, EGR1, USF1, CTCF, MAX, CREB1, ETS1, and FOS are most co-regulated TFs identified (Table S11).
Discussion
HPV is a pathogenic and infectious oncovirus causative agent of various cancers that pose a global burden of mortality [1, 4, 6]. One of the most important events in HPV infection and carcinogenesis is genomic integration, which alters host genes [12, 14, 18]. This reported contributing to aberrant proliferation, miRNA dysregulation, genomic instability, genomic structural alterations, cellular immortalization, epigenetic alterations, and malignant progression [12, 14, 53]. Disruption can also boost oncoprotein expression and lead to the loss of function of cell cycle checkpoints, DNA repair mechanisms, and tumor suppressor genes [14, 18, 54]. Therefore, recognizing potential druggable targets, molecular mechanisms, and predictive biomarkers is helpful for effective diagnosis and therapies.
In the study, we have utilized an integrative approach to identify key therapeutic targets concerning HPV pathogenesis, including CESC and HNSCC. Overall, 100 key target genes were identified out of 1887 candidates. Further, the role of these genes was explored in the HPVs pathogenesis and subjected to functional (GO) and pathway (KEGG) enrichment analyses. We have identified these are mainly related and correspond to the different binding activities to enzymes, receptors, and kinases, depicting the potential role in signaling, cell cycle, and DNA repair mechanisms. Further, we have also analyzed the TCGA-CESC and HNSCC data hosted at the GDC data portal and cBioPortal [20, 44, 46, 47] to further substantiate and prioritize the most promising druggable targets. This includes the genomic alterations, i.e., mutations (mainly missense and frameshift) and CNV (gain and loss).
Based on our analyses, 23 most promising therapeutic targets, i.e., TP53 (17p13.1), PIK3CA (3q26.32), NOTCH1 (9q34.3), EP300 (22q13.2), FN1 (2q35), CHEK1 (11q24.2), CUL1 (7q36.1), EZH2 (7q36.1), NRAS (1p13.2), H2AFX (11q23.3), SUMO1 (2q33.1), ATM (11q22.3), CREBBP (16p13.3), EGFR (7p11.2), ERBB2 (17q12), CCND1 (11q13.3), MYC (8q24.21), PTEN (10q23.31), KAT5 (11q13.1), RFC4 (3q27.3), PTK2 (8q24.3), RAD51 (15q15.1), and BUB1B (15q15.1) were identified. It is also noteworthy that these are coregulated and operated by different miRNAs including hsa-miR-155-5p, hsa-miR-34a-5p, hsa-miR-146a-5p, hsa-miR-203a-3p, hsa-miR-92a-3p, hsa-miR-193b-3p, hsa-miR-26a-5p, hsa-miR-145-5p, hsa-miR-24-3p, hsa-miR-199a-5p, hsa-miR-223-3p, hsa-miR-429, hsa-let-7a-5p, hsa-miR-30a-5p, hsa-miR-22-3p, hsa-miR-25-3p, hsa-miR-200c-3p, hsa-miR-125a-5p, hsa-miR-138-5p, and hsa-miR-451a found enriched in this study. Oncogenes (PIK3CA, NOTCH1, CREBBP, CCND1, NRAS, MYC, EGFR, ERBB2), transcription factors (TP53, CREBBP, EP300, EZH2, MYC, KAT5), protein kinases (CHEK1, ATM, EGFR, ERBB2, PTK2, BUB1B), tumor suppressors (TP53, EP300, ATM, PTEN, BUB1B), cell differentiation marker (ERBB2) are the most commonly altered genes among all.
These targets correspond to the multiple functions and pathways. Genomic alterations mainly occurred in G2M checkpoint genes (MYC, EZH2, H2AFX, CHEK1, CCND1, CUL1), E2F-Targets (MYC, EZH2, H2AFX, CHEK1, TP53, BUB1B), Apoptosis (CCND1, PTK2, ERBB2), TNFA signaling via NFKB (MYC, CCND1), PI3K_AKT_MTOR signaling pathways (EGFR, PTEN), WNT_BETA_CATENIN signaling (MYC, CUL1, TP53, NOTCH1), Apical junction (PTK2, EGFR, PTEN), Epithelial-mesenchymal transition (EMT) (FN1), P53 pathway (TP53, NOTCH1), DNA repair (TP53, CHEK1, RFC4, RAD51), COMPLEMENT system (FN1, PIK3CA) and NOTCH signaling pathways (CCND1, CUL1, NOTCH1). This also highlights the mechanisms disrupted or altered in the HPV pathogenesis. Different studies also report the misregulation of different pathways, mainly PI3K/Akt/mTOR signaling, Wnt/
Eight out of 23 potential targets are significantly down-regulated in both carcinomas. These are FN1 (25%, 17.9%), H2AFX (18%, 11.5%), CHEK1 (18.3%, 9.9%), ATM (15.8%, 10.7%), SUMO1 (15.5%, 11.3%), NRAS (9.9%, 13.8%), EZH2 (8.8%, 16.7%), and CUL1 (8.5%, 16.7%) along with affected case percentage for CESC and HNSCC, respectively. However, copy number loss of RAD51 (9.5%) and BUB1B (8.8%) are specific to CESC cases. Likewise, six promising targets, i.e., PIK3CA (20.1%, 27.2%), CCND1 (12.3%, 33.6%), RFC4 (19%, 25%), KAT5 (14.4%, 20.8%), MYC (13%, 15%), and PTK2 (11.6%, 11.5%) are upregulated in significant number of cases from both the carcinomas. While copy number gain of ERBB2 (8.1%) and EGFR (12%) are specific to CESC and HNSCC cases, respectively.
Different reports also suggest a potential role of many of these genes and miRNAs in various carcinomas, including CSCC and HNSCC. FN1 (Fibronectin 1) is an essential EMT component and plays a critical role in cell adhesion, growth, migration, differentiation, and metastasis. It is reported to be overexpressed and indicates poor prognosis in multiple cancer types, i.e., nasopharyngeal carcinoma [63, 64], gastric [65, 66, 67], colorectal [68], renal [69], oral squamous cell carcinoma (OSCC) [70], esophageal squamous cell carcinoma [71], thyroid [72], cervical [73] and HNSCC [74], etc. A study by Zhang et al. reported the miR-200c downregulation and FN1 upregulation in gastric cancer. The study explored the inhibitory effect of overexpression of miR-200c in gastric cancer [67]. Likewise, Dong et al. define the association of FN1 increased expression with the progression of clear cell renal cell carcinoma [75]. Further, Zhou et al. also demonstrate the upregulation of FN1 in cervical cancer and poor prognosis [73]. However, our study represents opposite trends with copy number loss (downregulation) of FN1 in the significant number of CESC and HNSCC cases. CHEK1 (checkpoint kinase 1) belongs to the Ser/Thr protein kinase family essential for the DNA damage-dependent cell cycle arrest. Al-kaabi et al. found a role of CHEK1 in breast cancer chemotherapy response, and nuclear expression is positively associated with favorable outcomes [76]. A study by van Harten et al. also reports CHEK1 as a promising therapeutic target in HNSCC and an important HNSCC cell cycle regulatory molecule [77]. Likewise, Yang et al. define the overexpression of CHEK1 in OSCC [78]. However, we have found a low copy number of CHEK1 in
Various high-throughput studies also advocated the use of specific genomic and transcriptomic applications and reported the crucial genes in CESC and HNSCC cases [13, 15, 18, 20]. Like, Ojesina et al. reported the somatic mutations in PIK3CA, PTEN, TP53, STK11, KRAS, MAPK1, EP300, FBXW7, NFE2L2, TP53, ERBB2 in CSCC and ELF3 and CBFB in adenocarcinoma [15]. Another study by The Cancer Genome Atlas Research Network defines the APOBEC, SHKBP1, ERBB3, CASP8, HLA-A, and TGFBR2 as novel mutated genes in 228 primary cervical cancers [20]. Likewise, Zhang et al. identify the differential expressed genes related to cervical intraepithelial neoplasia [21] and report enrichment of E2F-Targets, G2M-Checkpoint, Mitotic-Spindle, and Spermatogenesis pathways. Another study performed a meta-analysis of transcriptomics data. It revealed KAT2B, PCNA, CD86, PARP1, CDK1, GSK3B, WNK1, CRYAB, E2F4, ETS1, CUTL1, miRNAs (miR-192-5p, miR-193b-3p, and miR-215-5p), and some receptors like ephrin (EPHA4, EPHA5), endothelin (EDNRA, EDNRB) and nuclear (NCOA3, NR2C1, NR2C2) as potential biomarkers and target in cervical cancer [22]. Moreover, various studies also provide a mutational landscape from HNSCC. Stransky et al. analyzed whole-exome sequencing data and identified HNSCC mutated genes such as TP53, CDKN2A, PTEN, PIK3CA, HRAS, NOTCH1, IRF6, and TP63 [16]. Likewise, Seiwert et al. report the comparative analysis between HPV+ and HPV- HNSCC [88]. They show mutations in TP53, CDKN2A, MLL2, CUL3, NSD1, PIK3CA, and NOTCH in HPV- HNSCC. Also, mutant genes (FGFR2/3, DDX3X) and aberrations (PIK3CA, NOTCH1, KRAS, MLL2/3) in HPV+ HNSCC [88]. Likewise, Gaykalova et al. defined genetic alterations in TP53, NOTCH1, FGFR3, CEBPA, and FES [60].
To date, distinct clinical trials of five drugs for anogenital warts, human papillomavirus infection, and CIN have been conducted (ClinicalTrials.gov). These are IMIQUIMOD (CHEMBL1282) (Phase 2-4, completed) targeting Toll-like receptor 7 (TLR7), INTERFERON ALFA-2B (CHEMBL1201558) (Phase 2-4, completed) targeting interferon alpha and beta receptor subunit 1 and 2 (IFNAR1 and 2), INFIGRATINIB (CHEMBL1852688) (Phase 2, terminated) targeting Fibroblast growth factor receptor (FGFR) 1-4, CELECOXIB (CHEMBL118) (Phase 2, terminated) targeting prostaglandin-endoperoxide synthase 2 (PTGS2), and ALDESLEUKIN (CHEMBL1201438) (Phase 1, terminated) targeting interleukin 2 receptor subunit alpha and beta (IL2RA and IL2RB). However, clinical trials for two drugs were completed mainly for genital or perianal warts.
To substantiate our findings and approach, we have performed an extensive literature search to identify the existing drugs and targets shown to be effective in targeting HPVs and associated diseases. After this, we report (1) currently known active drugs and targets, (2) new repurposing drug candidates for known targets, and (3) novel targets and drug repurposing candidates. We have identified that anti-EGFR-targeted therapies are actively investigated with demonstrated benefits, including in clinical trials [29, 89, 90, 91]. It mainly includes monoclonal antibodies, i.e., cetuximab [34], nimotuzumab [92], panitumumab, zalutumumab, etc. and inhibitors such as gefitinib [93], erlotinib [94], lapatinib [95, 96], and dacomitinib [97]. In this study, we have also identified EGFR as one of the prominent targets with added repurposing therapeutic candidates, viz. imgatuzumab duligotuzumab, necitumumab, matuzumab, canertinib, brigatinib, osimertinib, pelitinib, pyrotinib, futuximab, tesevatinib, rociletinib, and olmutinib. Likewise, we found a few PI3K/mTOR inhibitors, namely dactolisib, alpelisib, and copanlisib, experimentally shown to abrogate HPV growth and tumor [98, 99, 100, 101]. We have identified understudied drug candidates (buparlisib, taselisib, gedatolisib, apitolisib, samotolisib, pictilisib, voxtalisib, serabelisib, sonolisib, pilaralisib) targeting PIK3CA, which could be further explored. A few studies show that ERBB2 targeting drugs, i.e., afatinib and neratinib, could be promising in inhibiting growth [101, 102, 103, 104]. We also report potential therapeutic regimens (ertumaxomab, trastuzumab, pertuzumab, margetuximab, gancotamab, CP-724714, tucatinib, poziotinib, sapitinib, varlitinib) as repurposing candidates targeting ERBB2 and pathways. Moreover, a few HDAC inhibitors, namely vorinostat, panobinostat, and belinostat, are effective therapeutic agents for treating HPV infections and carcinoma [93, 105]. We report added potential candidates, i.e., romidepsin, tacedinaline, entinostat, and mocetinostat, that could be used to target HPV+ carcinomas.
Further, we also found VEGFA as an important therapeutic target. A recombinant monoclonal antibody, i.e., bevacizumab, that inhibits VEGF was proven effective against HPV-associated diseases [35, 106, 107, 108]. We provide potential therapeutic regimens, namely vanucizumab, brolucizumab, and muparfostat, for repurposed clinical utility. We present AURKA and AURKB as effective targets for HPV-related cancers. Few reports also specify Aurora kinase (AURK) inhibitors, i.e., alisertib and danusertib, as promising approaches for treating cancers [109, 110]. We report understudied AURK inhibitors, namely tozasertib, barasertib, ilorasertib, and chiauranib, that could be explored further. Likewise, we also found CHEK1 as a promising target associated with HPV pathogenesis. Few drug repurposing candidates, i.e., prexasertib, rabusertib, and UCN-01, could be tested for anti-tumor activity against HPV-mediated carcinoma. Moreover, few studies have also shown the favorable outcome of inhibitor prexasertib against HPV-associated carcinoma [111, 112]. We have also found SRC as a promiscuous target and present dasatinib, vandetanib, tirbanibulin, bosutinib, and saracatinib for repurposing against HPVs. Similarly, PTK2 targeting drug molecules, namely defactinib, GSK-2256098, etc., could be clinically tested against HPV oncogenesis.
We have cataloged 230 potential drug candidates pertaining to key target genes identified in the study. These molecules could be further tested for the inhibition of HPV-associated infections. The results presented in our study may vary according to cohort and patients due to inherent heterogeneities in and between cancers, stages, and subtypes. Although we have performed integrative bioinformatics analyses to identify the most promising therapeutic targets and repurposed drug candidates, further experiments should be required for validation.
Conclusions
In summary, we have identified the critical target genes to counter HPV infections. An integrative approach is utilized, combining protein-protein interactome, functional and pathway enrichment, regulatory and TCGA cancer genomic data analysis. Overall, we have identified a number of promising druggable targets, including TP53, PIK3CA, NOTCH1, EP300, FN1, CHEK1, CUL1, EZH2, NRAS, H2AFX, SUMO1, ATM, CREBBP, EGFR, ERBB2, CCND1, MYC, PTEN, KAT5, RFC4, PTK2, RAD51, SRC, VEGFA, AURKA, AURKB, BUB1B, etc. against HPVs infections with cervical carcinoma and HNSCC. Likewise, significant transcriptional (TFs) and post-transcriptional (miRNAs) regulators/co-regulators were specified. This study also provides further research direction with respect to the potential biological pathways and molecular functions associated with HPVs pathogenesis. Importantly, we have cataloged potential drug repurposing candidates targeting distinct therapeutic targets. These targets and regulators may also serve as a potential prognostic biomarker for HPV-induced infections and carcinomas.
Author contributions
Conception: Manoj Kumar.
Interpretation or analysis of data: Amit Kumar Gupta.
Preparation of the manuscript: Amit Kumar Gupta.
Revision for important intellectual content: Amit Kumar Gupta and Manoj Kumar.
Supervision: Manoj Kumar.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210413.
sj-docx-1-cbm-10.3233_CBM-210413.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-210413.docx
Footnotes
Acknowledgments
This work was supported by the Council of Scientific and Industrial Research (CSIR), Government of India, grant number OLP0501, MLP0024. We would like to acknowledge Department of Biotechnology (DBT), Government of India [GAP0001] for infrastructure support. A.K.G. is supported by DST-INSPIRE fellowship sponsored by Department of Science & Technology (DST), Government of India.
Conflict of interest
The authors declare no conflict of interest.