
Abstract
BACKGROUND:
Melanoma is fatal cancer originating from melanocytes, whose high metastatic potential leads to an extremely poor prognosis.
OBJECTIVE:
This study aimed to reveal the relationship among EMT, TIICs, and immune checkpoints in melanoma.
METHODS:
Gene expression data and clinical data of melanoma were downloaded from TCGA, UCSC Xena and GEO databases. EMT-related DEGs were detected for risk score calculation. “ESTIMATE” and “xCell” were used for estimating TIICs and obtaining 64 immune cell subtypes, respectively. Moreover, we evaluated the relationship between the risk score and immune cell subtypes and immune checkpoints.
RESULTS:
Seven EMT-related genes were selected to establish a risk scoring system because of their integrated prognostic relevance. The results of GSEA revealed that most of the gene sets focused on immune-related pathways in the low-risk score group. The risk score was significantly correlated with the xCell score of some TIICs, which significantly affected the prognosis of melanoma. Patients with a low-risk score may be associated with a better response to ICI therapy.
CONCLUSION:
The individualized risk score could effectively conduct risk stratification, overall survival prediction, ICI therapy prediction, and TME judgment for patients with melanoma, which would be conducive to patients’ precise treatment.
Abbreviations
Introduction
Melanoma is fatal cancer originating from melanocy- tes, which comprises 65% of all skin cancers and whose high metastatic potential leads to an extremely poor prognosis [1, 2]. Several factors such as ultraviolet, radiation exposure, and malignant moles transformation contribute to melanoma formation [3]. In addition, the expression of oncogenes is associated with melanoma [4].
A previous study reported that the reactivation of epithelial-mesenchymal transition (EMT) was associated with the infiltration and metastasis of melanoma [5]. EMT promotes melanoma metastatic potential through epithelial trait loss and mesenchymal characteristics acquisition. In addition, the tumor microenvironment (TME) that is associated with tumor growth and metastasis has a relationship with tumor prognosis, including melanoma. Moreover, a strong association had been reported between EMT status and tumor immune microenvironment in melanoma [6, 7]. EMT-related differentially expressed genes (DEGs) in the TME are involved in regulating the function of tumor infiltration immune cells (TIICs), which has direct killing effects on tumor cells that may be the reason of tumor progression and metastasis.
A recent study demonstrated that programmed cell death protein 1 (PD-1) therapy-resistant patients with melanoma present distinct signatures of upregulated genes involved in monocyte chemotaxis and EMT [8]. The extracranial activation/release of CD8
In this study, we constructed a risk model on the basis of the Cancer Genome Atlas (TCGA) cohort and validated it using the GSE65904 cohort. The risk model is based on seven EMT-related genes (ESRP1, FBP1, JAG2, NME1, PIM2, PSME3, and STAT1) which were found to be strongly associated with CD8
Patients and methods
Microarray data acquisition
We downloaded gene expression data and clinical data of skin cutaneous melanoma from the TCGA database (
Identification of DEGs and PPI network construction
“Estimate” package (version 1.0.13) R language (version 4.0.0) was used to estimate the ratio of the immune-stromal component in TME for each sample. TME results included the immune and stromal scores. The higher the respective score is, the larger the ratio of the corresponding component in TME would be. The EMT-related DEGs were obtained by using the R software “limma” package according to the parameters
Establishment of a prognostic gene signature for melanoma
Univariate Cox proportional hazards regression analysis was used to identify prognostic relevant genes from EMT-related DEGs. Moreover, Kaplan-Meier plots and log-rank tests were adopted to validate the relationship between the DEGs and overall survival. The 20 out of the 28 DEGs obtained from the Cox regression and Kaplan-Meier analyses were further used for LASSO regression analysis to select the genes included in the model. Risk scores were calculated using the generated coefficients and corresponding expression. Patients were divided into “high-risk” and “low-risk” groups on the basis of the median risk score obtained using this prognostic model. To further assess the diagnostic value of the risk score system, time-dependent receiver operating characteristics (ROC) and survival analysis were performed.
Gene set enrichment analysis (GSEA)
Hallmarks (h.all.v72.symbols.gmt) and c2 (c2.cp.kegg.v7.2.symbols.gmt) gene sets were downloaded from the Molecular Signatures Database [12] as target sets, using which gene set enrichment analysis (GSEA; V4.0.3) was performed between high-risk and low-risk score groups. The significant cutoff value was defined as the false discovery rate of
xCell score estimation
The “xCell” package was used to calculate the cell type enrichment score (xCell scores) based on the gene expression data. Notably, the xCell tool provides 64 cell types, including lymphoid, myeloid, stromal cells, stem cells, and other cells. The difference between each cell subtype between the high- and low-risk score groups was compared using the
Relationships between risk score, immune cells and immune checkpoints
Pearson’s correlation analysis between the risk score and the xCell score of immunity cells was performed using the “corrplot” package (version 0.84). Pearson’s correlation coefficient of
Construction of nomograms
The nomograms were constructed using patients with melanoma in TCGA dataset. Moreover, univariate and multivariate Cox proportional hazards regression analyses were performed on the risk score of EMT-related genes signature and other clinical characteristics. Hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated. Nomograms were constructed in this study using information obtained from the results of multivariate Cox regression analysis. The predictive accuracy of the nomogram was assessed by ROC curve analysis.
Immunohistochemistry
Patients volunteering for the study had signed informed consent forms. In addition, this study was approved by the Ethics Department of the First Affiliated Hospital of Guangxi Medical University and conformed to the World Medical Association Declaration of Helsinki. Six pairs (melanoma and paracancerous tissues) of pathological sections for each gene used for immunohistochemistry analysis. The PSME3 and NME1 antibodies for immunohistochemical staining were purchased from the ABclonal. The PIM2, JAG2 and ESRP1antibodies were purchased from the Abmart. The FBP1 and STAT1 antibodies were purchased from the Abcam. After paraffin removal, hydration, and sealing, the specimens were mixed with anti-ESRP1, -FBP1, -JAG2, -NME1, -PIM2, -PSME3, and -STAT1 and incubated overnight at 4
Univariate Cox regression analyses to the DEGs
Univariate Cox regression analyses to the DEGs
EMT-related DEGs were mainly involved in inflammation response
“The raw score obtained from “Estimate” package for the TCGA cohort was shown in supplementary file S1. Patients had a better prognosis in the high immune score group than the low immune score group (Fig. 1A). However, patients with a high stromal score had no effect on prolonging the prognosis (Fig. 1B). A total of 801 DEGs (168 upregulated and 633 downregulated) were identified in melanoma compared with the normal skin (Fig. 1C). In addition, 256 DEGs (135 upregulated and 121 downregulated) were identified in the high immune score group compared with the low immune score group (Fig. 1D). Moreover, 297 DEGs (228 upregulated and 69 downregulated) were identified in the metastatic melanoma compared with nonmetastatic melanoma (Fig. 1E). The common DEGs (22 up-regulated and 6 down-regulated) shared by melanoma, immune score, and metastatic status were selected for further analysis (Fig. 1F and G). The PPI network is shown in Fig. 1H. GO and KEGG enrichment analyses showed that DEGs were mainly involved in inflammation response, including T-cell activation and response to chemokine and cytokine (Supplementary Fig. S1A, S1B; Supplementary File S2).
Correlation of Immune status and the prognosis in melanoma and DEGs identification. (A) Kaplan-Meier survival curve for the immune score with 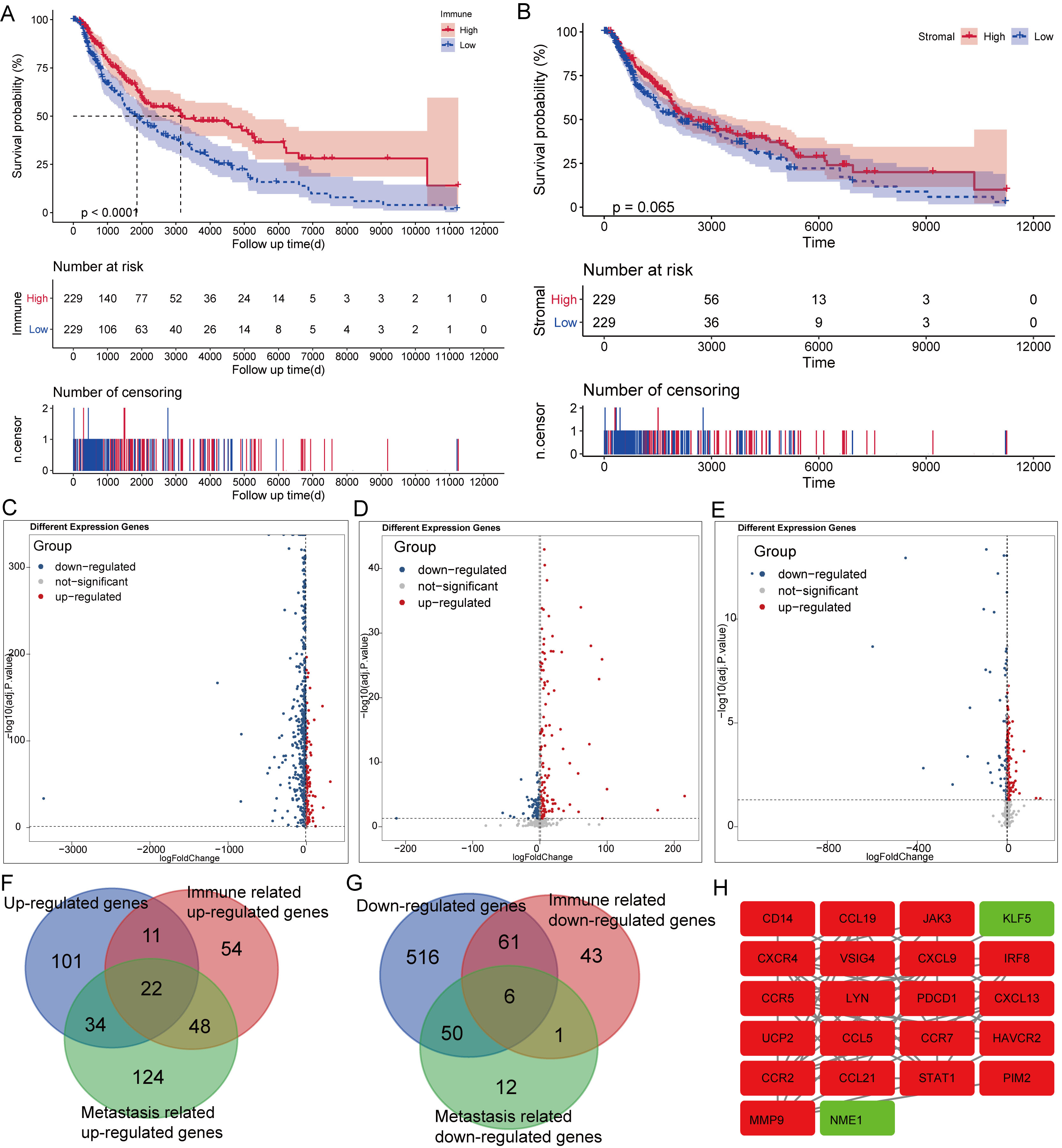
Establishment of a prognostic gene signature for melanoma. (A, B) 7 of the DEGs selected by LASSO Cox regression analysis. Left: using 1000-fold cross-validation to the optimal penalty parameter lambda. Right: LASSO coefficient profiles of the 20 DEGs. (C) Distribution of the risk score in the TCGA cohort. Upper panel: classification of patients into different risk groups based on the median risk score. Middle panel: distribution of patients’ survival time and status. Bottom panel: Heatmap of expression profiles of included risk score related genes. (D) Kaplan-Meier survival curves between low- and high-risk groups. (E, F) Validation of the risk score system prognostic signature in the GSE65904.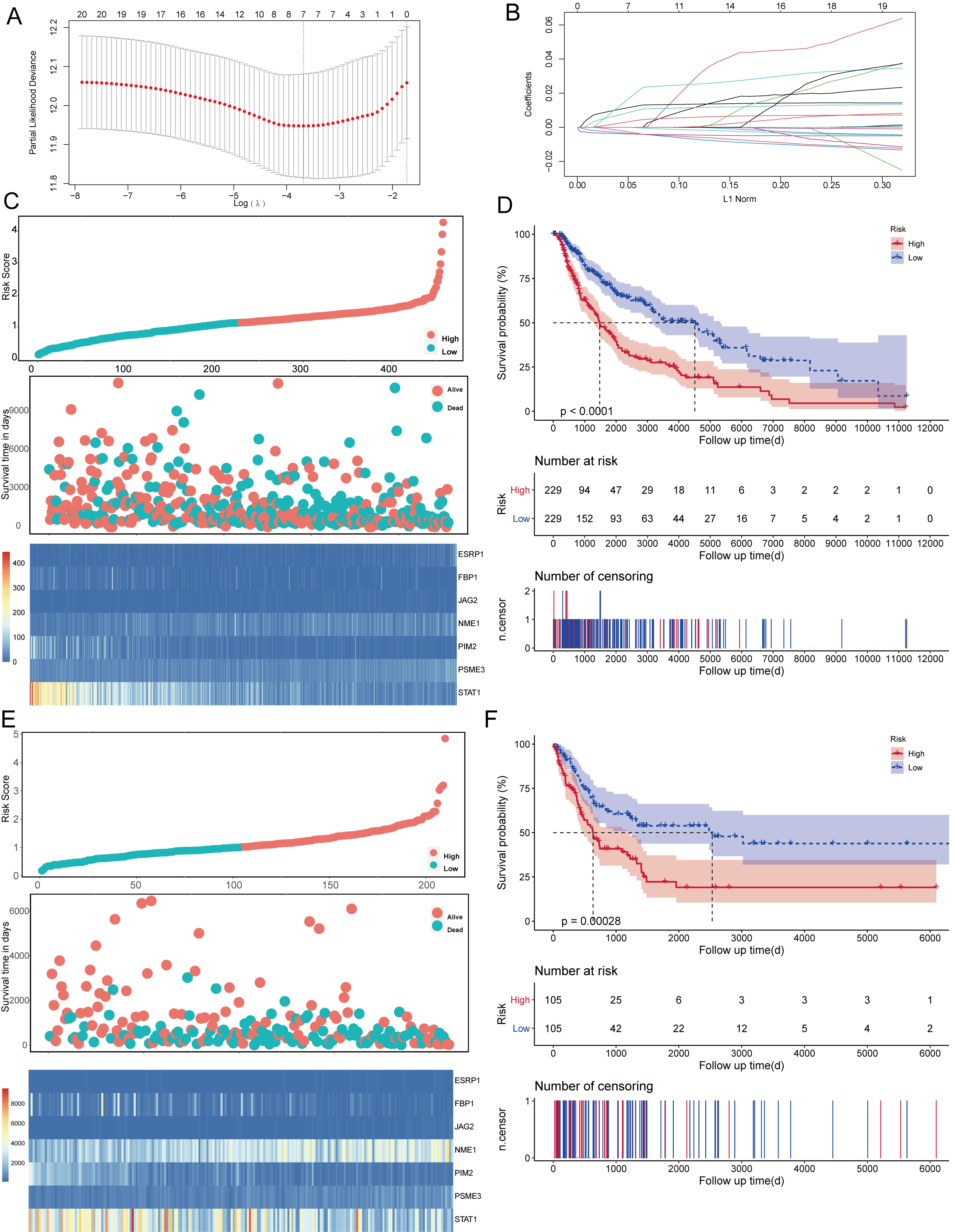
GSEA analysis. (A) GSEA in the Hallmarks (left) and KEGG (right) gene set between group high (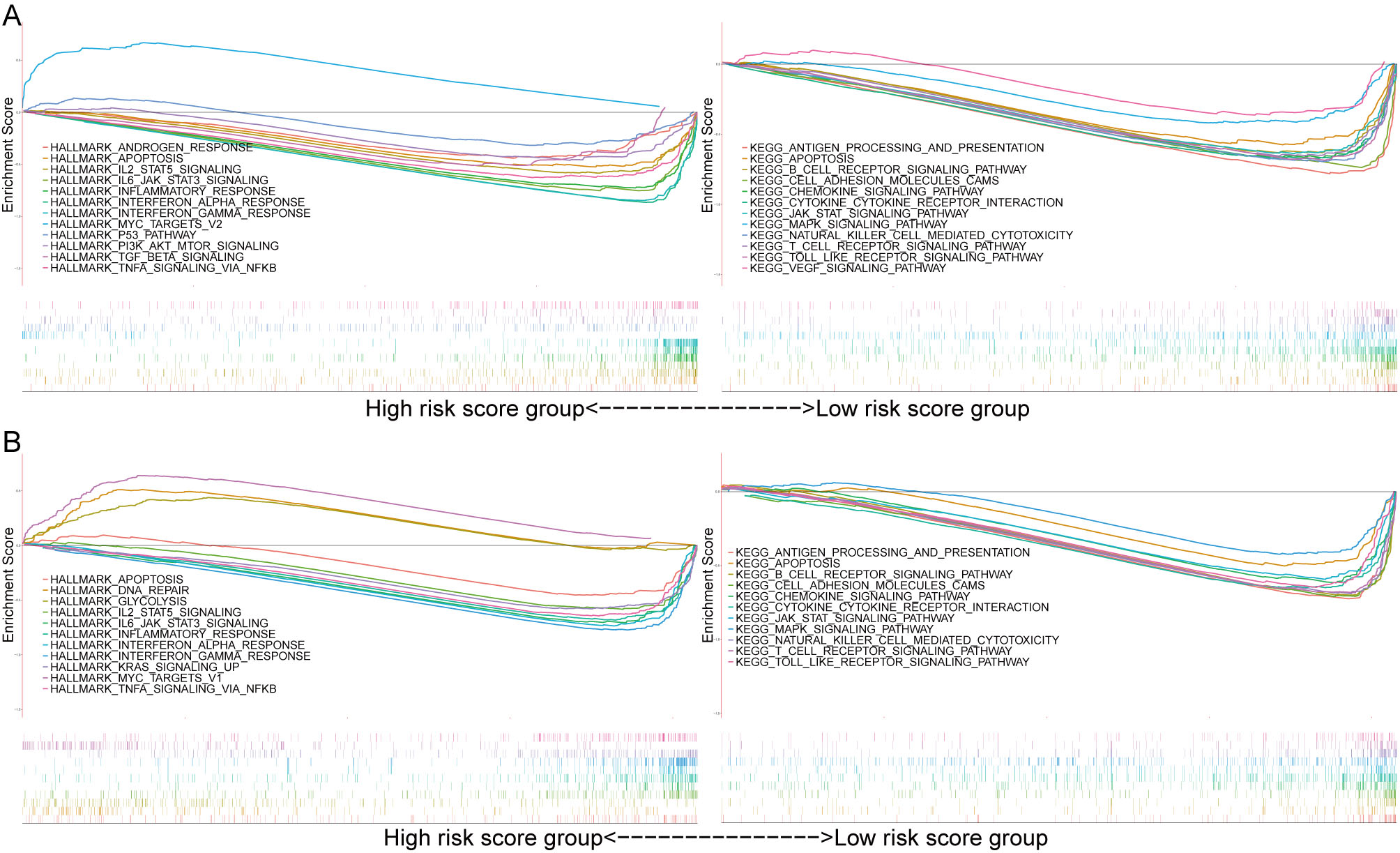
A total of 21 out of 28 DEGs were screened as the significant prognosis factors from the univariate Cox regression analysis (Table 1). Moreover, 24 out of 28 DEGs affected melanoma prognosis as noted from Kaplan-Meier analysis (Supplementary Fig. S2A). The 20 out of 28 DEGs shared by Cox regression analysis and Kaplan-Meier analysis (Supplementary Fig. S2B) were further used to perform LASSO regression analysis for selecting the genes included in the model (Fig. 2A and B). A seven EMT-related DEGs (ESRP1, FBP1, JAG2, NME1, PIM2, PSME3, and STAT1) risk signature was established. All these seven DEGs were significantly differently expressed between high- and low-risk score groups in the TCGA (Supplementary Fig. S3A) and GSE65904 cohort (Supplementary Fig. S3B). On the basis of the median risk score, patients were separated into two groups, high- and low-risk groups, wherein the high-risk group had lower survival rates (Fig. 2C and D). The GSE65904 cohort, which was used to validate the prognostic model, showed similar results (Fig. 2E and F).
The risk score predicts the involvement of immune pathways
A large number of genes were enriched in immunity- and tumor-related pathways. Results from the GSEA analysis were similar to GO and KEGG enrichment analyses to the DEGs. In the TCGA cohort, the Hallmark background showed enrichment mainly for apoptosis, IL2/STAT5 signaling, IL6/JAK/STAT3 signaling, inflammatory response, interferon
TIICs profile in different risk groups
The lymphoid and myeloid cells were considerably lower in the high-risk score group in the TCGA cohort (Supplementary Fig. S4A, S4B, S4C, S4D, S4E) and GSE65904 cohort (Supplementary Fig. S5A, S5B, S5C, S5D, S5E). Three subtypes of immunity cells were significantly upregulated, whereas 25 subtypes of immunity cells were significantly downregulated in the high-risk score group (Supplementary Fig. S5F). Pearson’s correlation analysis showed that 14 and eight subtypes of TIICs were significantly correlated with the risk score in the TCGA (Fig. 4A) and GSE65904 cohorts (Fig. 4B), respectively. The Venn plot showed that activated dendritic cells (aDC), pDC, CD8
The correlation of risk score with xCell score. (A) The correlation between the risk score and xCell score in the TCGA cohort. (B) The correlation between the risk score and xCell score in the GSE65904 cohort.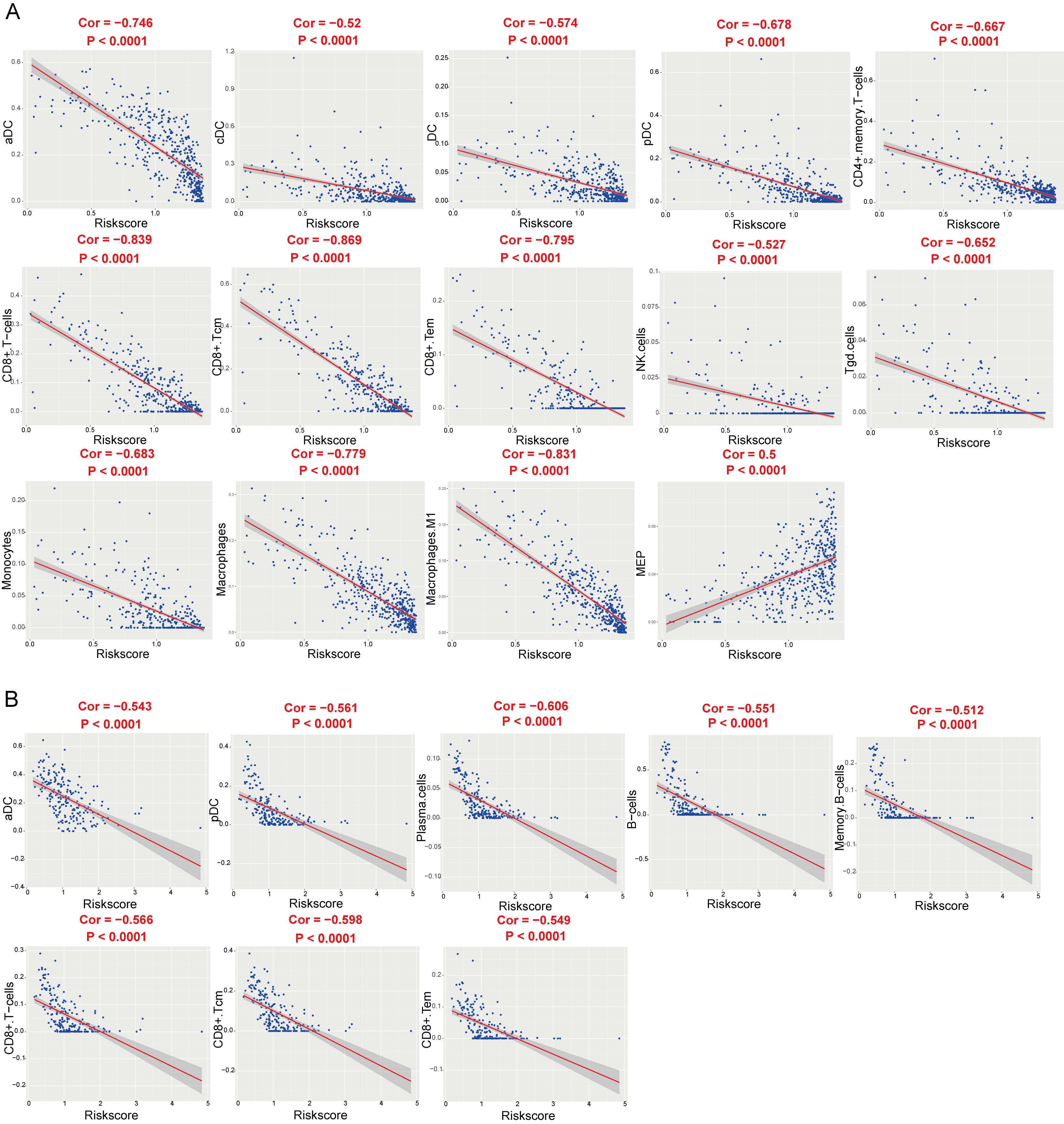
Kaplan-Meier survival analysis to the immunity cells. (A) Venn plot showing the immunity cells that was significantly correlated with the risk score in the TCGA cohort and the GSE65904 cohort. Kaplan-Meier survival curves between low- and high-aDC(B), pDC(C), CD8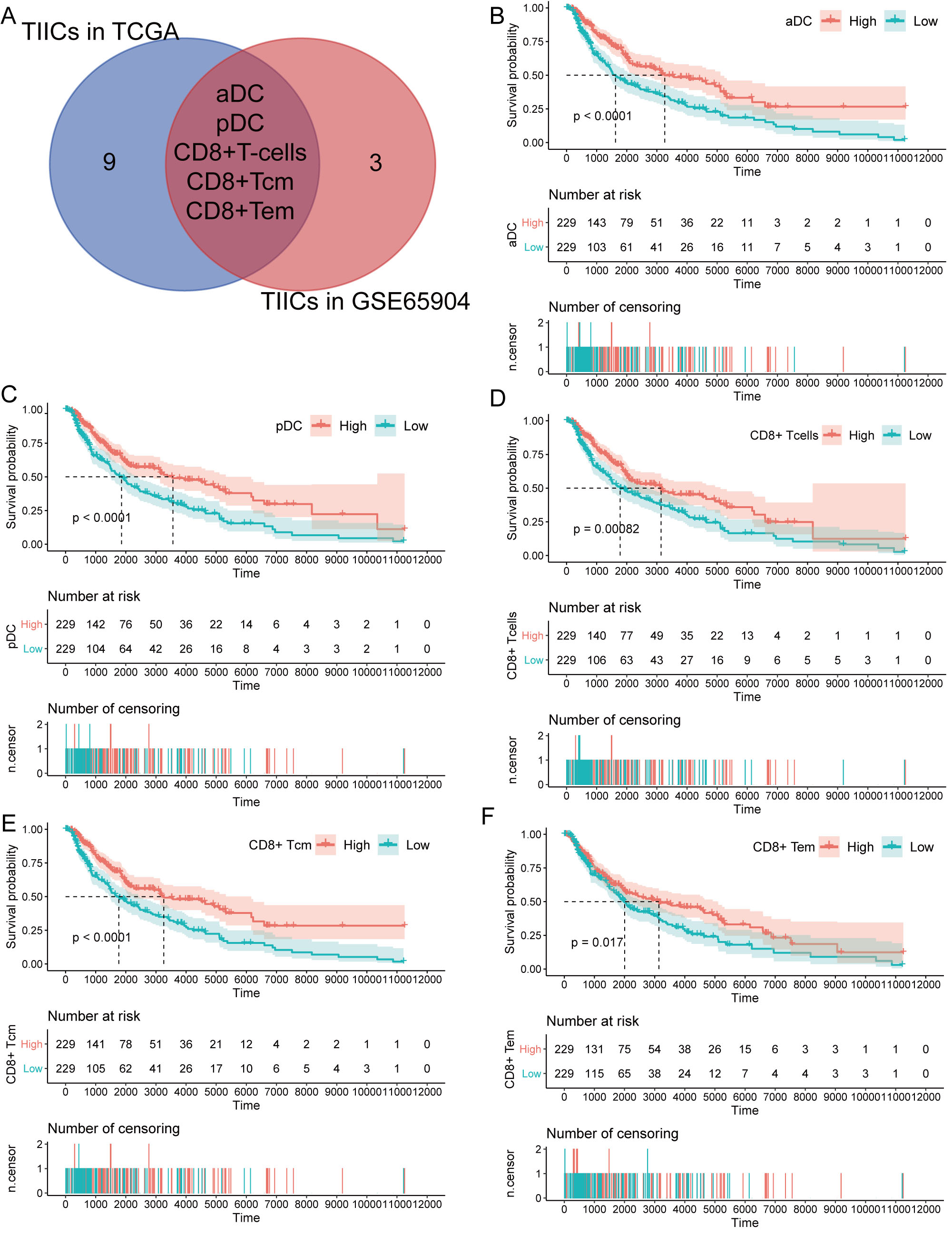
Kaplan-Meier survival curves between low- and high-aDC(A), pDC(B), CD8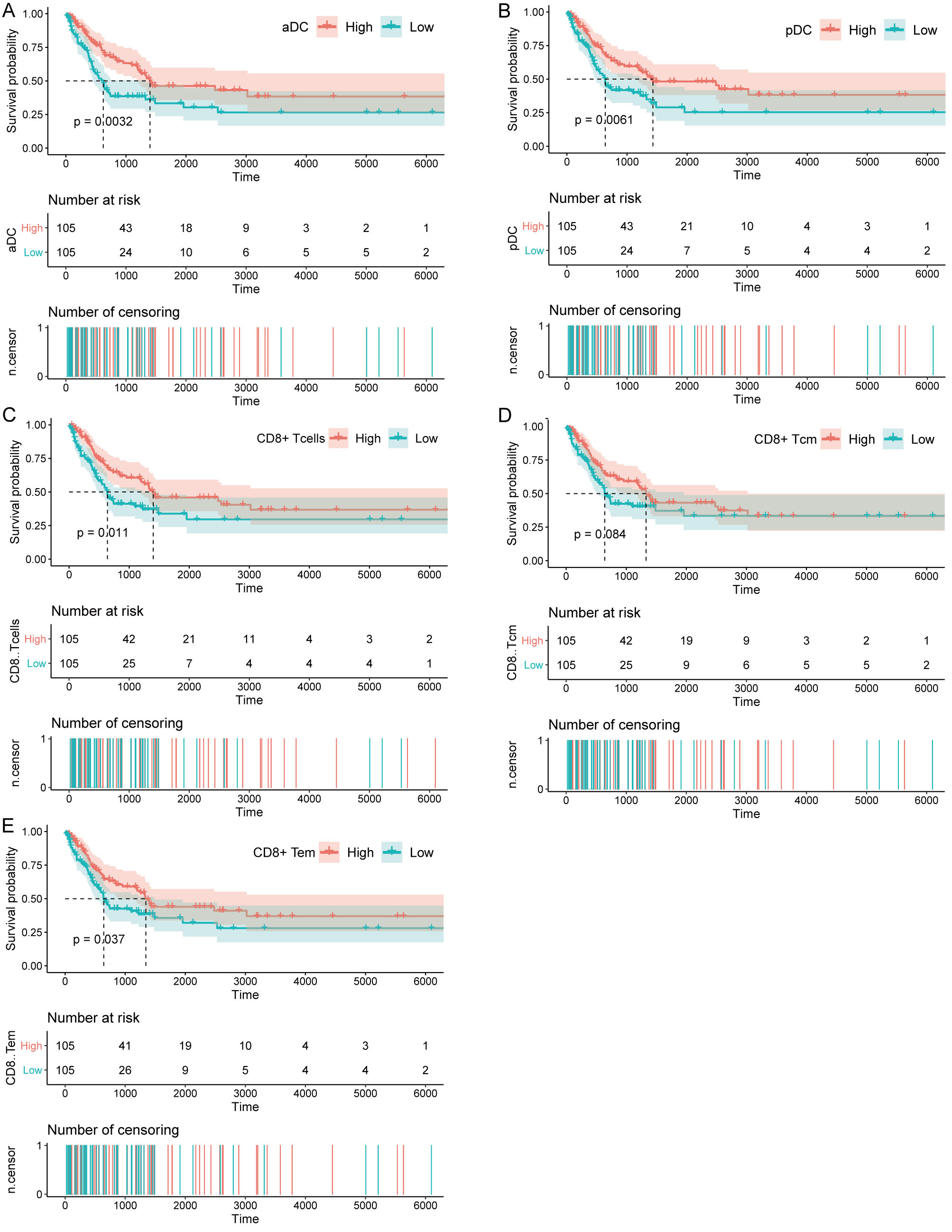
The expression levels of PD-1, PD-L1, and CTLA-4 were significantly higher in the low-risk score group than the high-risk score group in the TCGA (Fig. 7A) and GSE65904 cohorts (Fig. 7B). Furthermore, patients with a low-risk score had a better survival outcome than those with a high-risk score in the TCGA (Fig. 7C, E, G) and GSE65904 cohorts (Fig. 7D, F, H).
Impact of immune checkpoint gene expression and EMT-related genes signature on clinical outcome. Comparison of the expression pattern of immune checkpoint genes (PD1, PD-L1, and CTLA-4) between high-risk score group and low-risk score group in the TCGA cohort (A), GSE65904 cohort (B). Kaplan-Meier survival curves of OS among four patient groups stratified by the risk score and PD-1, PD-L1 and CTLA-4 in the TCGA cohort (C, E, G) and GSE65904 cohort (D, F, H).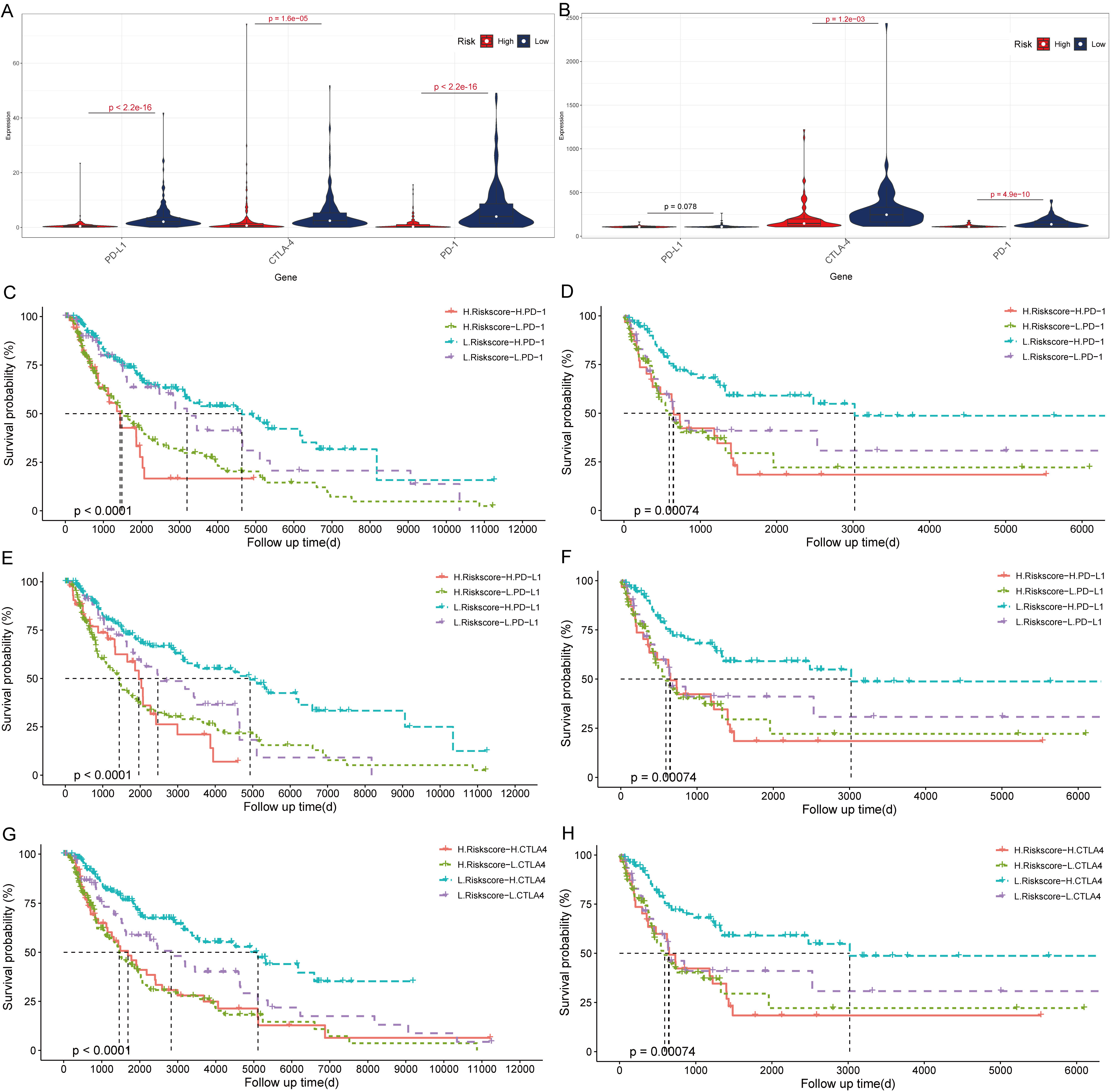
Age, race, T stage, N stage, and risk score were incorporated in the nomogram (Table 2). Moreover, a total point summarized the points of each variable, predicting the probability of overall survival at 1, 3, and 5 years (Fig. 8A). The area under curve (AUC) at 1, 3, and 5 years in the nomogram model showed higher accuracy (AUC
Univariate and multivariate Cox analysis of TCGA cohort
Univariate and multivariate Cox analysis of TCGA cohort
Nomogram and correlation analysis. (A) Nomogram for predicting the probability of overall survival at 1, 3 and 5 years. (B) The 1, 3 and 5 years AUC of the nomogram model. (C) The 1, 3 and 5 years calibration plots of the nomogram.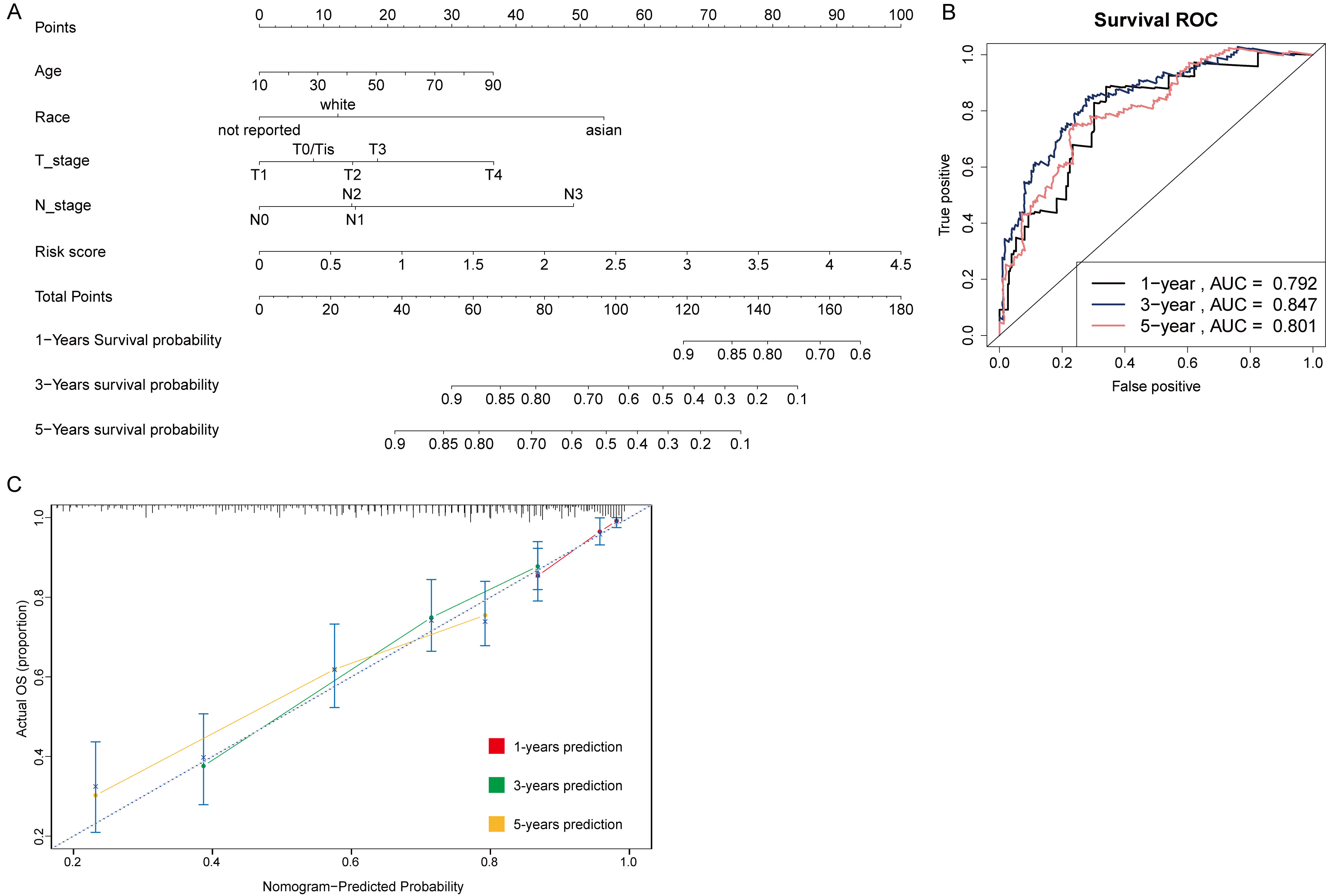
Immunohistochemical plots of the three hub genes associated with prognosis and statistical analysis of the positivity rate. (A, C, E) shows the protein expression of each gene in melanoma and in the paracancerous tissue. (B, D, F) shows the statistical analysis of the staining positivity rate for each gene in melanoma and in the paracancerous tissue. 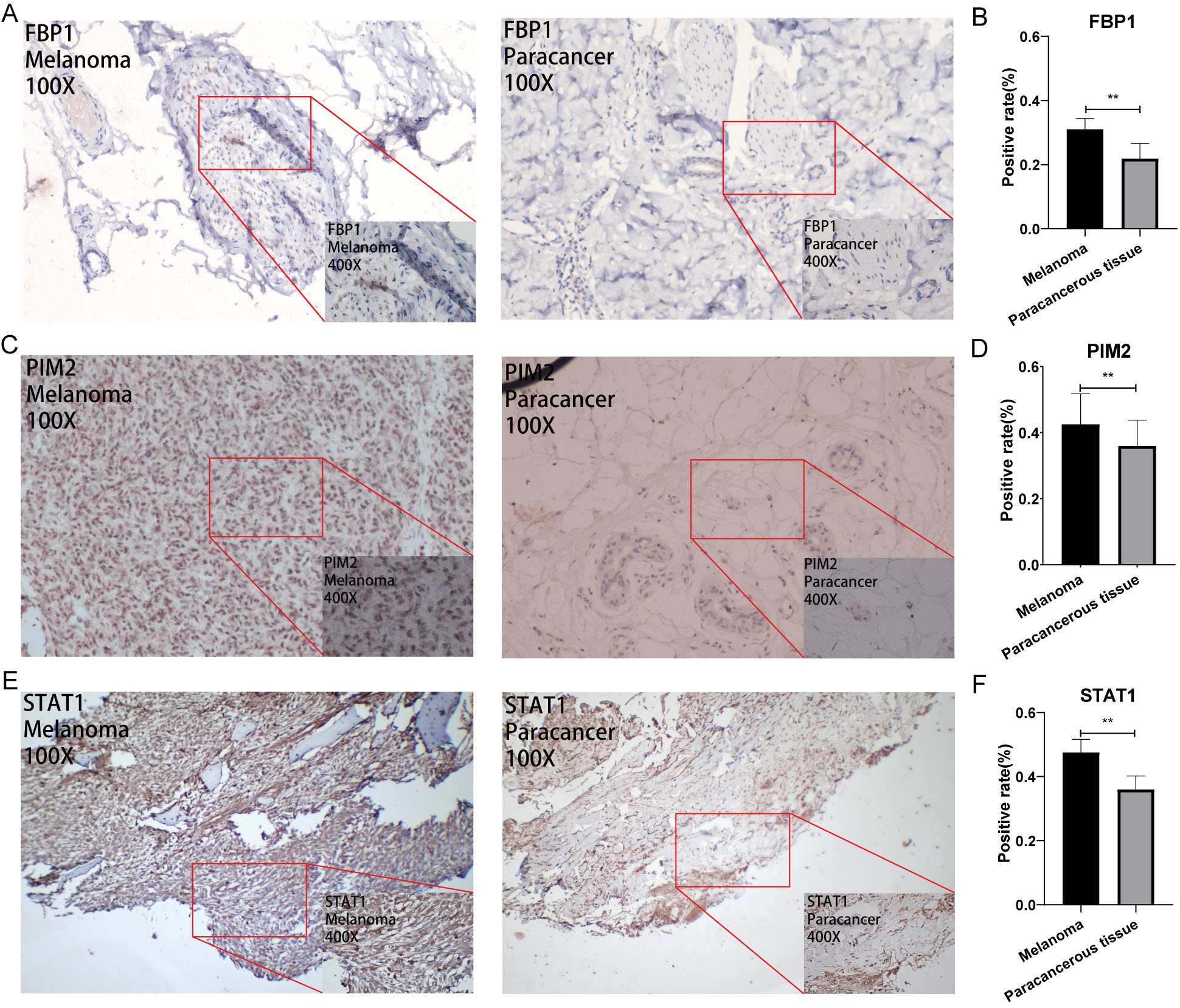
Immunohistochemical plots of the four hub genes associated with prognosis and statistical analysis of the positivity rate. (A, C, E, G) shows the protein expression of each gene in melanoma and in the paracancerous tissue. (B, D, F, H) shows the statistical analysis of the staining positivity rate for each gene in melanoma and in the paracancerous tissue. 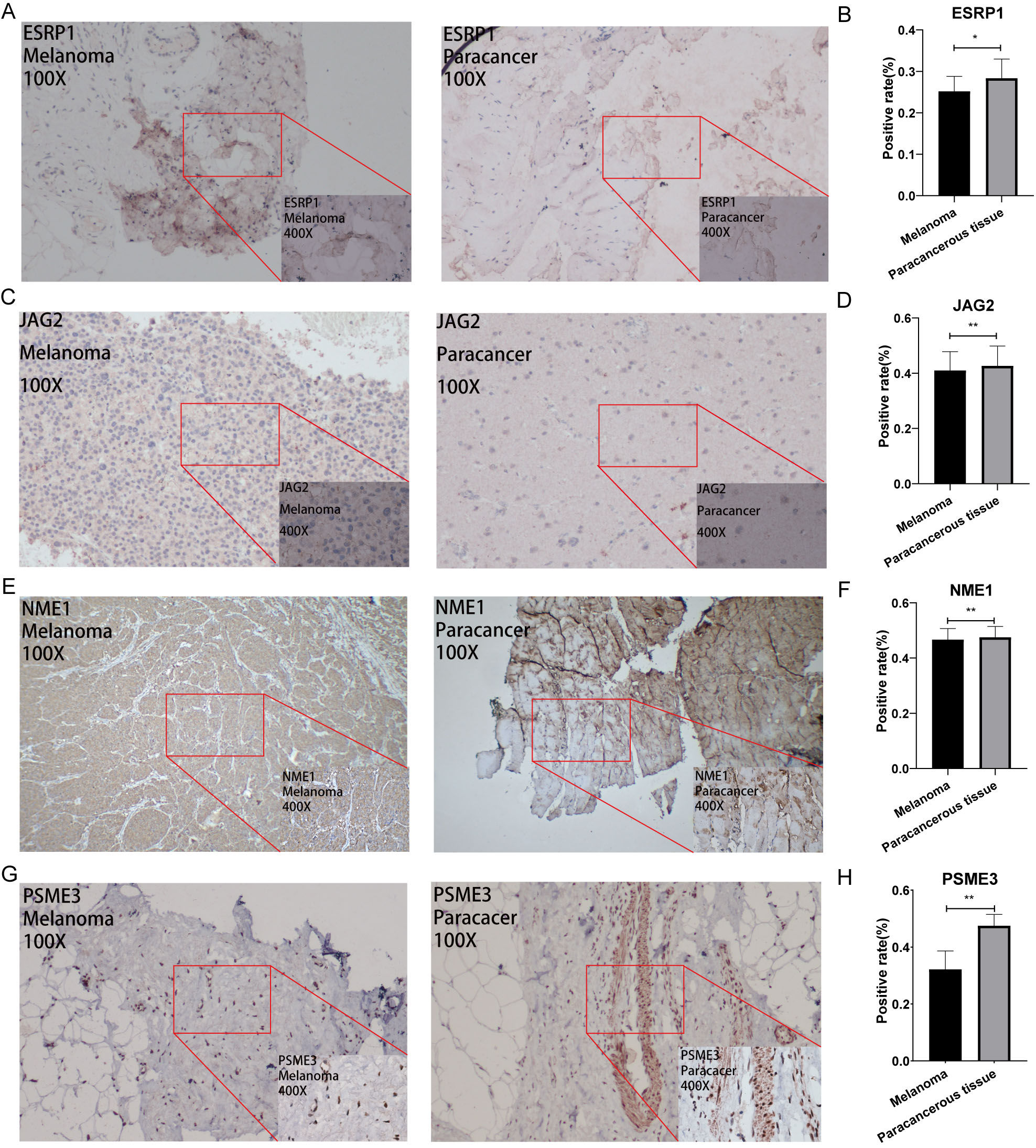
All immunohistological images were observed under an inverted microscope and images were collected. Moreover, the staining differences between osteosarcoma and paraneoplastic tissue specimens were compared. The FBP1, STAT1, and PIM2 antibodies were significantly upregulated, whereas ESRP1, JAG2, NME1, and PSME3 were significantly downregulated in melanoma compared with that in paraneoplastic tissue. Representative pictures are showed in Figs 9 and 10. In addition, Fig. 9B, D, and F showed that the positive rate of immunohistochemical staining for FBP1, STAT1, and PIM2 was significantly higher in melanoma than that in paraneoplastic tissues. Fig. 10B, D, F, and H showed that the positive rate of immunohistochemical staining for ESRP1, JAG2, NME1, and PSME3 was significantly lower in melanoma than that in paraneoplastic tissues.
Discussion
The normal melanocytes can transition to metastatic melanomas because of EMT [14]. Moreover, EMT, which was reported to correlate with the tumor immune microenvironment, including the lymphoid and myeloid cells compartment, leads to antitumor and tumor-promoting effects by shifting the balance of the immune system [15]. EMT plays a crucial role in tumor progression because of the metastatic transition and immune system dysregulation.
We identified 28 EMT-related DEGs associated with immunity and metastasis to further reveal the relationship between EMT and tumor metastasis and TME. Finally, seven EMT-related genes out of the 28 DEGs were screened to establish the risk model, and the GSE65904 cohort was used for validation. The risk model showed a good prognosis and diagnosis predictive ability for melanoma. Moreover, the seven EMT-related genes that were incorporated in the risk model were associated with melanoma and its metastatic: Epithelial Splicing Regulatory Protein 1 (ESRP1), Signal Transducer and Activator of Transcription 1 (STAT1), Pim-2 Proto-Oncogene (PIM2), Jagged Canonical Notch Ligand 2 (JAG2), Fructose-Bisphosphatase 1 (FBP1), NM23 Nucleoside Diphosphate Kinase 1 (NME1), and Proteasome Activator Subunit 3 (PSME3). Moreover, JAG2 and NME1 can promote the expansion of cells with enhanced tumor and metastatic potential in the context of melanoma cell cultures [16, 17], whereas, FBP1, PIM2, and STAT1 act as a tumor suppressor in melanoma [18, 19, 20]. In addition, ESRP1 can work as informative biomarkers for immunotherapy because of the regulation ability of TIICs in melanoma [21]. Our results consolidated previous data that the EMT is strongly associated with melanoma and its metastatic potential.
Subsequently, we analyzed the relationship between EMT and TIICs in the TME. The results showed that three subtypes were downregulated, and 25 subtypes were upregulated in the low-risk group. The infiltration of immune cells, including aDC, pDC, CD8
PD-1, PD-L1, and CTLA-4, the common immune checkpoint molecules [26, 27, 28], were associated with the progression and prognosis of melanoma. ICI therapy has enormous potential in improving clinical outcomes for several tumors, including melanoma [29]. PD-1, PD-L1, and CTLA-4 are currently available biomarkers in clinical practice but not a sufficient independent predictor of ICI response [30]. Therefore, it is crucial to find predictive biomarkers that could identify patients who are most likely to benefit from ICI. In this study, the expression of immune checkpoint genes was much higher in the low-risk group. The survival analysis was performed for patients divided into four groups on the basis of the risk score and the immune checkpoint expression level. Moreover, we found that patients with melanoma at low risk and high immune checkpoint expression level have a better prognosis. These results indicated that ICI therapy might be more effective in patients in the low-risk group. Moreover, a previous study reported that ESRP1 can work as informative biomarkers for immunotherapy because its downregulation may promote the probability of responding to immune checkpoint blockade therapy to melanoma [31]. In addition, STAT1, an upstream gene in the pathway “PD-L1 expression and PD-1 checkpoint pathway in cancer”, was strongly associated with PD-L1 expression. These findings thus revealed that the risk model could work as a predictor for ICI response.
GSEA analysis was performed to further reveal the molecular mechanism between high- and low-risk groups. GSEA analysis showed that inflammatory response and immune-related pathways such as IL2/STAT5 signaling, interferon
Conclusion
A novel EMT-related risk score was developed and validated in an independent cohort. The individualized risk score could effectively conduct risk stratification, overall survival prediction, ICI therapy prediction, and immune microenvironment judgment for patients with melanoma, which would be conducive to patients’ precise treatment.
Declarations
Consent for publication
Not applicable.
Ethics approval and consent to participate
All subjects volunteered for the study and signed informed consent forms. In order to ensure confidentiality, the names of study participants were not included in the data. Information obtained from the data of the study participants is kept confidential. In addition, the Ethics Committee of The First Affiliated Hospital of Guangxi Medical University approved the study.
Data availability statement
The dataset from the TCGA database generated and/or analyzed during the current study are available in the TCGA dataset repository (
Competing interest
We declare that we do not have any competing interests.
Funding
This work was sponsored by the National Natural Science Foundation of China (81560359); National Natural Science Foundation of China (81860393). Funding bodies had no role in the study design, collection, analysis, and interpretation of the data or in writing the manuscript.
Author contributions
Zhan Xinli, Liang Tuo: Conceptualization, Methodology. Chen Jiarui, Xu GuoYong, Zhang Zide: Data curation, Investigation. Xue Jiang, Zeng Haopeng, Jiang Jie: Formal analysis, Software. Qin Zhaojie, Hao Li, Chen Tianyou, Ye Zhen, Nie Yunfeng: Visualization. Liang Tuo, Liu Chong: Writing- Reviewing and Editing.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210329.
sj-xlsx-2-cbm-10.3233_CBM-210329.xlsx - Supplemental material
Supplemental material, sj-xlsx-2-cbm-10.3233_CBM-210329.xlsx
sj-xlsx-1-cbm-10.3233_CBM-210329.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-210329.xlsx
sj-pdf-1-cbm-10.3233_CBM-210329.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-210329.pdf
Footnotes
Acknowledgments
We are grateful to Dr. Xinli Zhan (Spine and Osteopathy Ward, The First Affiliated Hospital of Guangxi Medical University) for his kindly assistance in all stages of the present study.