
Abstract
BACKGROUND:
Fatty acid oxidation has been considered as an important energy source for tumorigenesis and development. Several studies have investigated the role of CPT1A, a kind of fatty acid oxidation rate-limiting enzyme, in AML. However, prognostic value and regulatory network of another subtype, CPT1B in AML remains elusive. This study aims to clarify the independent prognostic role of CPT1B in CN-AML based on clinical data and molecular level data (mRNA, miRNA and lncRNA).
OBJECTIVE:
The aim of this study is to investigate the prognostic value of CPT1B in AML patients.
METHODS:
First, we analyzed the CPT1B expression in AML cohort via the online database “GEPIA”. Subsequently, miRNA-mRNA and ceRNA networks were constructed to help predict the role of CPT1B in AML. Several molecules which showed the prognostic value and metabolic function of CPT1B were identified. Finally, the expression of CPT1B in our own cohort of 324 CN-AML patients was analyzed to clarify the results.
RESULTS:
It was found that CPT1B was markedly higher in AML patients compared to normal people and this upregulation was associated with the poor clinical outcome. Several molecules revealed the possible regulatory mechanism of CPT1B in AML.
CONCLUSION:
CPT1B is a potential prognostic factor and a therapeutic target for AML treatment.
Background
Acute myelocytic leukemia (AML) is a group of hematological malignancies with high heterogeneity in terms of cell morphology, molecular biology, immunotyping, and cytogenetics. This disease is characterized by high mortality, high treatment-related mortality, high relapse rates and poor clinical outcomes [1]. Currently, there are no molecular markers to assist personalized therapy. Studies have shown that chromosomal abnormalities can be used to effectively stratify the risk of AML.However, chromosomal abnormalities were shown as an effective tool for AML risk stratification but cytogenetically normal acute myeloid leukemia (CN-AML) accounts for 50% of all types of AML and constitutes the main body of intermediate-risk AML with heterogeneous groups [2]. Molecular diagnosis of NPM1, FLT3-ITD and CEBPA mutation analysis have been applied in risk stratification of CN-AML patients [3]. Since CN-AML patients have specific cytogenetic consistency without abnormalities in their chromosomes, they provide a perfect platform for identification of AML biomarkers. Molecular signals such as DNA mutations, abnormal mRNA and miRNA expression can be favorable or unfavorable disease biomarkers.
Changes in metabolic signals have been linked to the occurrence of leukemia, hence can be exploited to develop treatments for AML [4]. Traditionally, the Warburg effect of aerobic glycolysis was considered to be an important source of bioenergy for cell proliferation [5]. Recent studies have shown that fatty acid oxidation (FAO) can also be an important energy source for cancer pathogenesis [6]. The carnitine palmitoyltransferase 1A (CPT1A), a subtype of CPT1, potentially catalyzes the rate-limiting step in FAO. This enzyme has been found to be an independent risk factor that predicts poor AML prognosis [7]. A study by Ricciardi et al. showed that CPT1A inhibitor (ST1326) could block FAO thereby exert antileukemic effects based on leukemia cell lines and primary cells obtained from patients with hematologic malignancies [8]. Majority of studies on CPT1B have been limited to fatty acid metabolism and only a few have investigated its role in tumors. Previous studies showed that the STAT3-CPT1B-FAO pathway promotes breast cancer progression and the emergence of chemoresistance [9]. Thus, CPT1B may be an important prognostic and therapeutic target.
These results highlight the tumor-promoting role of CPT1B in AML. However, the clinical relevance of CPT1B in AML as well as its regulatory mechanisms are unclear.
Herein, results showed that CPT1B could be an important prognostic predictor of AML. The identified unique mRNA, miRNA, lncRNA patterns consequently decipher the biologic insights of high CPT1B expressors. Unlike previous studies on CPT1B, the present study investigated the effect and mechanism of CPT1B in AML using bioinformatics and clinical data.
Methods
Clinical patients
The AML patients involved in this study were all diagnosed at the age of 14 and above. Bone marrow specimens used for genetic analyses were obtained from each patient at the time of diagnosis prior to any treatment. All these patients were diagnosed with CN-AML. Clinical data of 324 AML patients were abstracted from medical records between January 2010 and July 2016. As previously described, the WHO classification, cytogenetic and molecular analysis were done centrally at the Zhejiang Institute of Hematology (ZIH) in China [10]. Using methods as previously described, chromosomal abnormalities were analyzed and RT-qPCR was conducted to assess mutations of genes such as DNMT3A, NPM1, CEBPA, FLT3-ITD, IDH1 and IDH2 [11]. The above cytogenetics analysis were performed by researchers without knowing the CPT1B expression level and clinical outcome. All patients provided written informed consent to participate. This study was approved by the Research Ethics Committee of the First Af?liated Hospital, College of Medicine, Zhejiang University.
Quantitative reverse transcriptase-PCR
The process of RNA extraction, reverse transcription and quantitative PCR has been reported [12]. Quantification was based on
CPT1B, 5
Statistical analysis
Clinical data analysis
Patient characteristics were summarized by descriptive statistics, including frequency counts, median, and range. The main purpose of this study was to clarify the prognostic value of CPT1B expression in AML patients as well as the possible upstream and downstream regulatory molecules and pathways of CPT1B in AML. Overall survival (OS) is defined as the time from the date of diagnosis to death due to any causes. The definition of event free survival (EFS) was time from date of diagnosis to removal from the study because of the absence of complete remission (CR), relapse or death. Adjusted variables such as age, WBC, DNMT3A gene, IDH1 and IDH2 mutations were considered to be recognized indicators for AML patients. The association between CPT1B expression and OS, EFS was assessed by the Kaplan-Meier method and log-rank test. The Fisher exact and Wilcoxon rank-sum tests were used, respectively for categorical and continuous variables, so as to assess the association between expression levels and clinical molecular characteristics. The impacts of CPT1B expression to OS and EFS were evaluated via multivariable hazards models in the presence of other known risk factors.
Sequencing data analysis
Expression profiles of mRNAs, miRNAs, lncRNAs and clinical information about 68 CN-AML patients were obtained by high throughput sequencing (RNA-Seq), derived from the Cancer Genome Atlas (TCGA) (
mRNA analysis
Protein protein interaction network (PPI network) was developed using STRING [13] and visualized by Cytoscape software [14]. The top30 hub genes analysis was done by the APP named “cytohubba” [15]. Hallmarks analysis was done by Gene Set Enrichment Analysis [16]. The data of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was obtained from STRING before visualized via Cytoscape software.
miRNA analysis
The KEGG pathways enrichment for those DE miRNAs were performed via mirPath v3 [17]. Interaction of miRNAs and mRNAs integrative analysis were done by WGCNA via R statistic packages, version3.6.3 [18].
ceRNA network construction
The co-expression network of differentially expressed mRNAs, miRNAs and lncRNAs were constructed to ascertain the roles of mRNAs, miRNAs and lncRNAs in mediated ceRNA network. DE miRNAs targeting mRNAs were retrieved from miRTarBase (
Results
Association of CPT1B overexpression with poor clinical outcome in TCGA Cohort
With the analysis of public data sets in the GEPIA database, we explored the potential of CPT1B as an independent prognostic factor. CPT1B expression was associated with poor OS [HR: 2.6,
Survival curves of AML patients. a: High CPT1B expression is associated with poor AML OS (Analysis by online database GEPIA). b: CPT1B expression in AML patient is significantly higher than it in normal people (red presents AML patient while grey presents normal people). 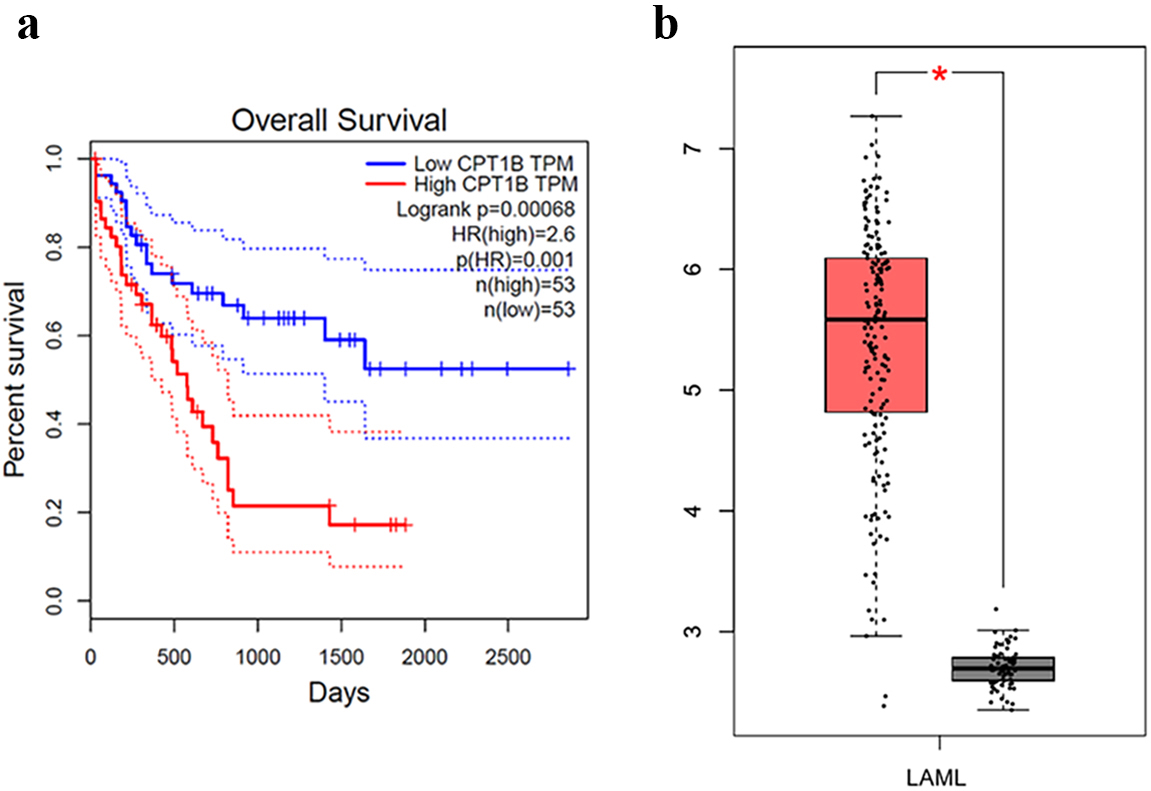
In the TCGA cohort, a meta-analysis of gene expression profiles was conducted in 68 CN-AML patients to explore the biological role of CPT1B in leukemogenesis. A total of 10 pairs were obtained by matching CN-AML patients with high and low CPT1B expression based on variables like age (less than 1 year old), sex (the same sex), WBC and genes including FLT3-ITD, NPM1, CEBPA, DNMT3A in TCGA cohort (Table S1). The 10 pairs were further subjected to differential analysis. The results showed a total of 413 were up-regulated and 164 were down-regulated genes in the TCGA cohort (Table S2). The constructed PPI network via online data STRING (Table S3) was used to determine the interaction between proteins. The top 30 hub genes were determined using via cytoHubba in both up-regulated and down-regulated differential genes (Fig. 2A–B, Table S4).
Associations between genome-wide expression profiles and CPT1B expression. a: Top30 hub genes in up-regulated differential genes. b: Top30 hub genes in down-regulated differential genes. c: Differential hallmarks between high and low expression of CPT1B. d: Top30 KEGG pathways about up-regulated hub genes. e: Top30 KEGG pathways about down-regulated hub genes. 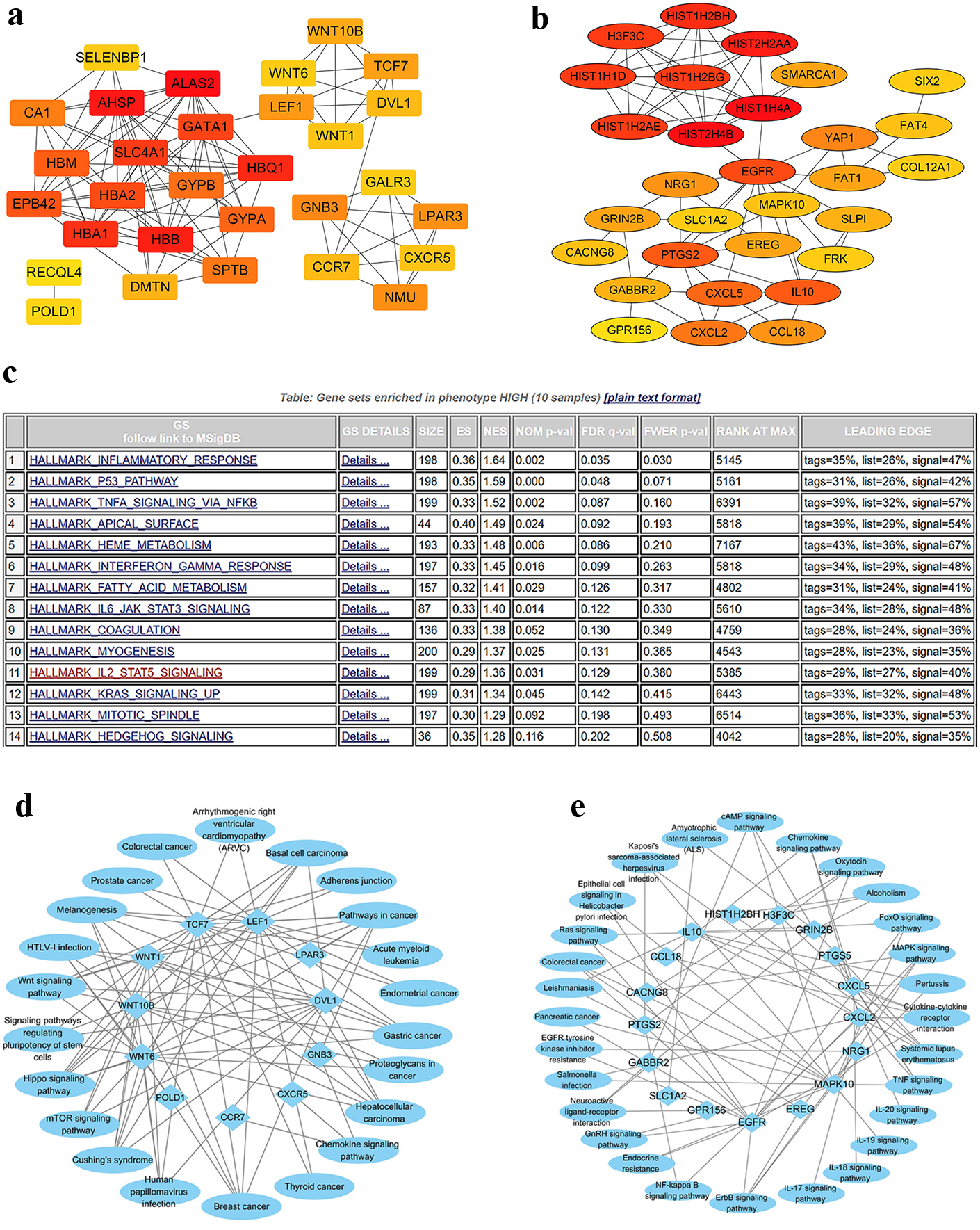
In the TCGA data, differential pathways between CPT1B high and low expressors were analyzed using the GSEA method. There were upregulated hallmarks in CPT1B high expressors such as inflammatory response, P53 pathway, TNFA signaling via NFKB, fatty acid metabolism, IL6-JAK-STAT3, IL2-STAT5, KRAS signaling, and others (Fig. 2C). Most of these pathways were reported in AML. KEGG pathways enrichment analysis was particularly for the hub genes to elect the top30 pathways among the up-regulated and down-regulated hub genes, respectively (Fig. 2D–E). The up-regulated hub genes were associated with several pathways such as acute myeloid leukemia, Wnt signaling pathway, mTOR signaling pathway whereas the down-regulated hub genes were linked to NF-KB signaling, MAPK signaling among other pathways.
According to KEGG results of hub genes, LEF1 (lymphoid enhancer-binding factor 1) was elected since its high expression serves as a novel favorable prognostic factor in cytogenetically normal acute myeloid leukemia [19]. However, ALAS2, an erythroid-specific mitochondrially located enzyme, potentially predict the OS outcome in AML [20]. Therefore, their genes and associated pathways can provide enormous knowledge of CPT1B.
The aforementioned criteria for matching the CN-AML patients with high and low CPT1B expression in the TCGA cohort (Table S1) was adopted whereby the differential miRNAs were further evaluated based on 10 pairs obtained. Finally, 48 downregulated and 82 upregulated miRNAs were identified in high CPT1B expression groups (Table S5).
KEGG pathways enrichment analyses were performed to identify pathways associated with the DE miRNAs via mirPath v3. It was found the upregulated DE miRNAs were linked to the fatty acid metabolism, fatty acid elongation, citrate cycle (TCA cycle), cell cycle, p53 signaling pathway, TNF signaling pathway, mTOR signaling pathway, chronic myeloid leukemia, and acute myeloid leukemia pathways. In contrast, the down-regulated DE miRNAs were linked to TGF-
KEGG analysis of disregulated miRNAs. a: KEGG analysis of up-regulated miRNAs. b: KEGG analysis of down-regulated miRNAs. 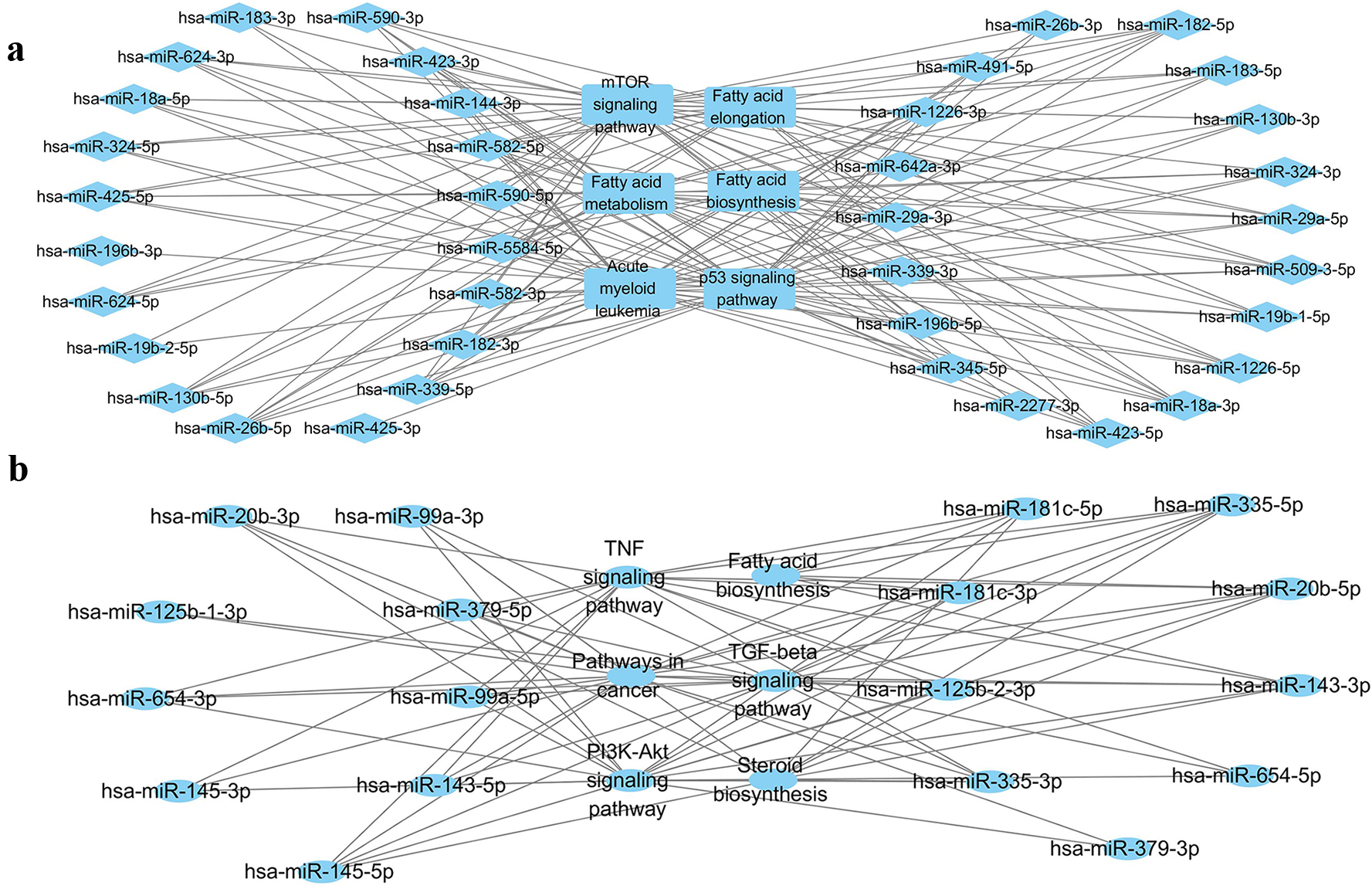
The results of KEGG pathways enrichment for DE mRNAs and DE miRNAs pointed out a certain direction for our subsequent research. By combining the KEGG results of miRNAs and mRNAs, we revealed that the role of CPT1B was likely related to P53, TNF, mTOR signaling pathway and several other metabolism processes.
The WGCNA (abline picks 0.6) tool was used to analyze the interactions between mRNA and miRNA in high and low CPT1B expression groups. Five downregulated genes in high CPT1B expression group were predicted to be targeted by the upregulated miR-509.3. Similarly, 2 downregulated genes were likely to be targeted by upregulated miR-339,etc (Fig. 4A, Table S7).
miRNA-mRNA network. a: Network of up-regulated miRNAs and their targeted down-regulated mRNAs in high CPT1B expressor (Red represents up-regulated miRNAs while blue represents down-regulated mRNAs). b: High expression of miR-339 is associated with poor survival of AML. 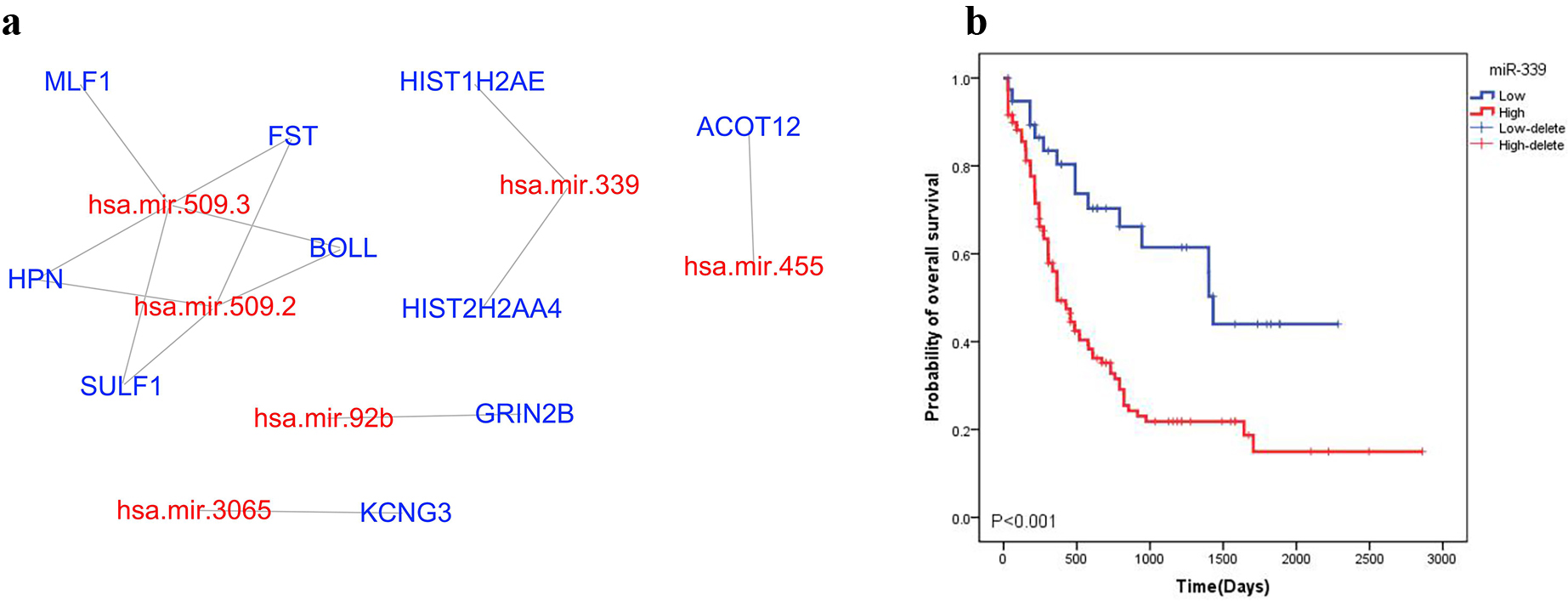
ceRNA network. a: ceRNA net about up-regulated differiential miRNA and its targeted lncRNA and mRNA. b: ceRNA net about down-regulated differiential miRNA and its targeted lncRNA and mRNA. 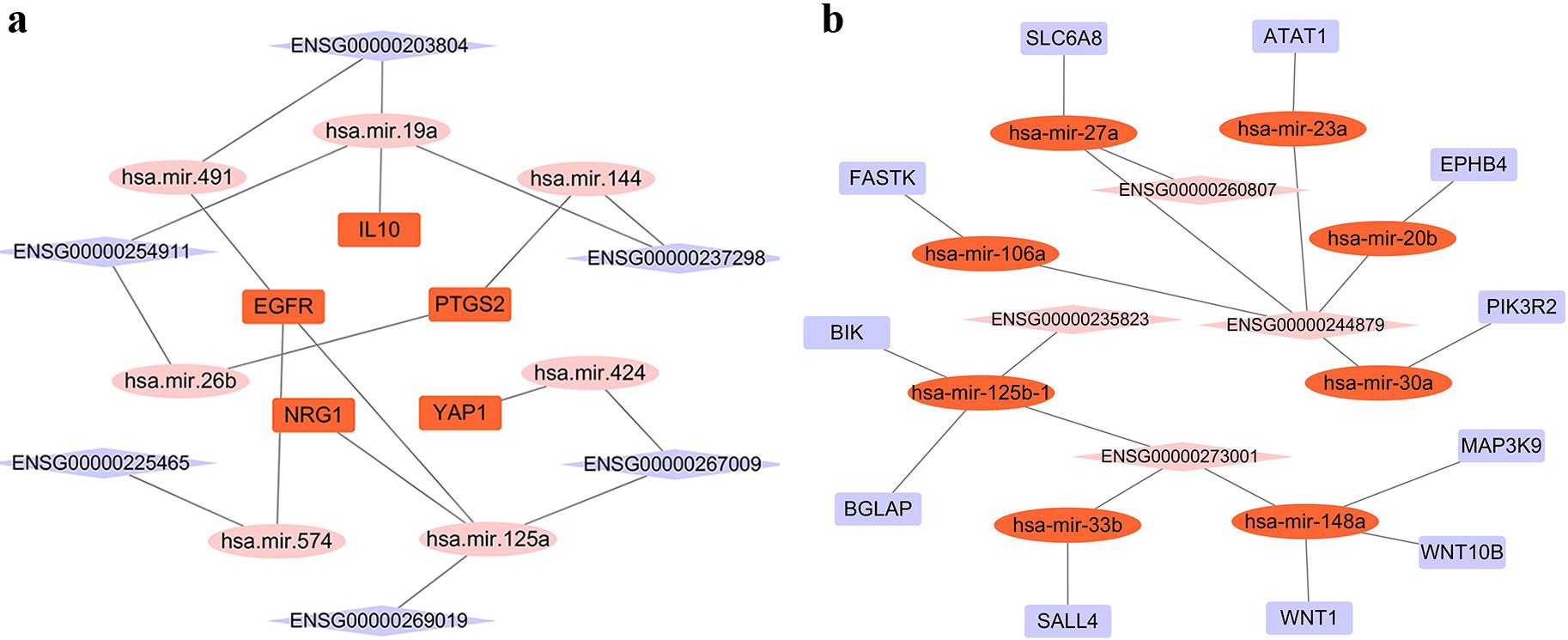
Next, we investigated the biological processes and KEGG pathways enrichment associated with the dysregulated mRNAs and miRNAs in the miRNA-mRNA network. In the BP process, miR-509.3 was linked to FST and MLF1 genes in the hematopoietic progenitor cell differentiation, HPN and SULF1 genes in the negative regulation of epithelial cell proliferation and regulate BOLL in the positive regulation of translational initiation process. The miR-455 was predicted to regulate ACOT12 gene in acetyl-CoA metabolic process (Table S8). In the KEGG pathways enrichment analysis, miR-509.3 was associated with FST in the TGF-beta signaling pathway whereas miR-339 was linked to HIST2H2AA4 and HIST1H2AE in the necroptosis. The miR-455 was predicted to regulate ACOT12 gene in pyruvate metabolism (Table S9). This analysis indicate that some miRNA/mRNA are involved in the biological processes in high CPT1B expression groups. Among the miRNAs, the high expression of miR-339 was associated with poor survival of AML (Fig. 4B). Furthermore, ACOT12 (acyl-CoA thioesterase 12) was associated with the metabolic processes suggesting the metabolic function of CPT1B.
Prognosis of dysregulated lncRNAs. a: Upregulated of ENSG00000235823 predicts poor outcome. b: Upregulated of ENSG00000273001 predicts poor outcome. c: Downregulated of ENSG00000203804 predicts poor outcome. d: Downregulated of ENSG00000237298 predicts poor outcome. e: Downregulated of ENSG00000267009 predicts poor outcome. f: Downregulated of ENSG00000269019 predicts poor outcome.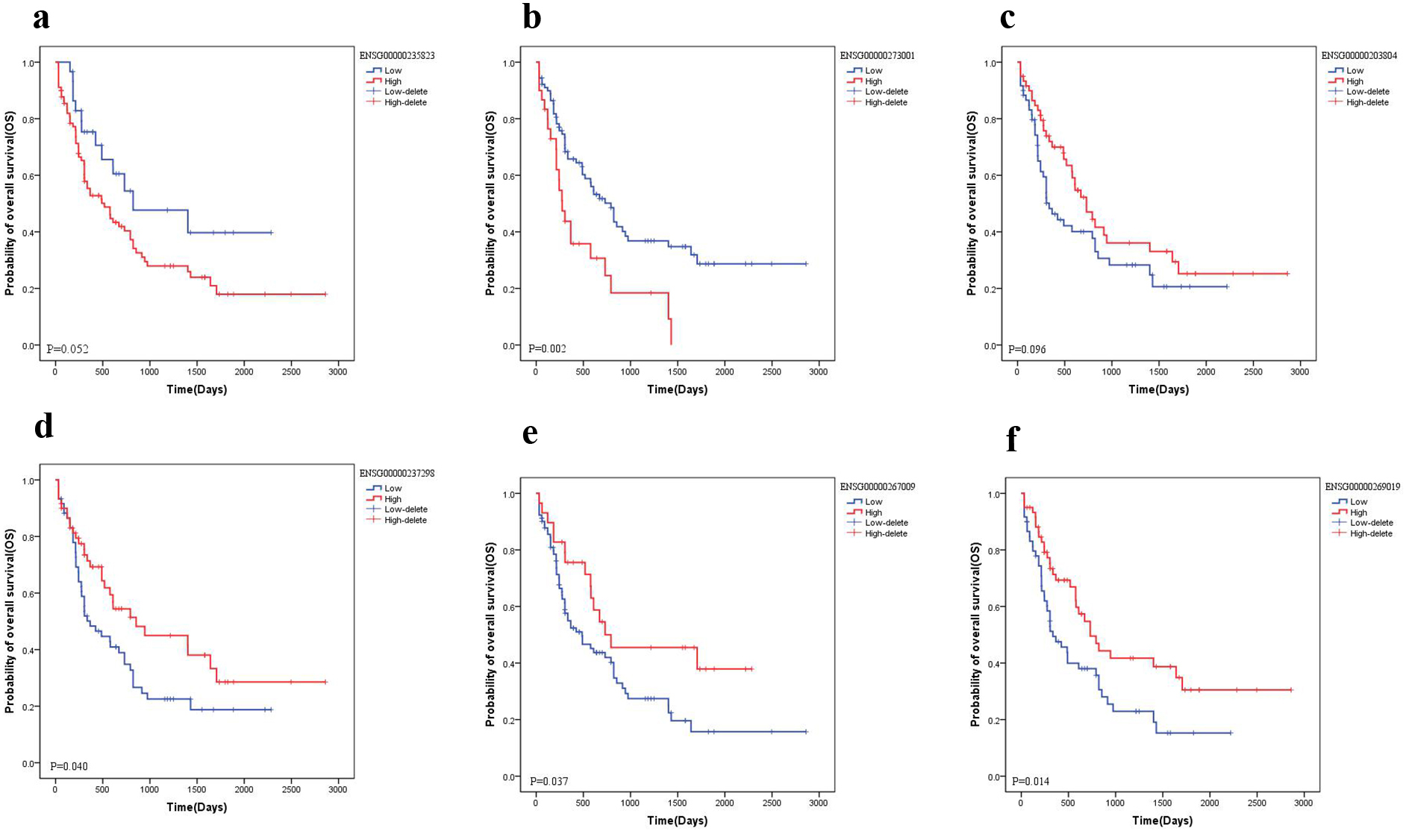
Long non-coding RNAs (lncRNAs) have a considerable influence on tumorigenesis. To investigate the function of lncRNAs in different CPT1B expressors, we conducted the meta-analysis on lncRNA expression profiles of 68 CN-AML patients based on the TCGA cohort. Patients matching and their analysis were conducted as aforementioned (Table S1). From the analysis, 116 up-regulated and 59 down-regulated lncRNAs were identified (Table S10).
The lncRNAs were predicted using differential miRNAs based on online database LncBase-v2 before intersected with the DE lncRNAs that was earlier analyzed. In this process, we identified 28 up-regulated miRNAs and 14 down-regulated lncRNAs as well as 15 down-regulated miRNAs and 10 up-regulated lncRNAs (Table S11). The 28 up-regulated miRNAs and 15 down-regulated miRNAs were used to predict mRNAs based on online data mirTarBase. The predicted mRNAs were intersected with the pre-analyzed DE mRNAs. In this process, we should ensure: 1. mRNA and lncRNA are positively correlated 2. mRNA and lncRNA have a common related miRNA, and they are negatively correlated with miRNA expression, finally 14 dysregulated miRNAs and 10 dysregulated lncRNAs, as well as 16 dysregulated mRNAs, were found for ceRNA construction. Consequently, a ceRNA network was constructed based on the above data and visualized using Cytoscape v3. 7. 1 (Fig. 5) to provide insight into the mediation mechanisms of mRNA-miRNA combination.
Prognosis of lncRNA in ceRNA network
To dig out the significant lncRNAs, we made prognosis analysis of the differential lncRNAs in ceRNA network. There were several lncRNAs associated with the prognosis of AML. For the up-regulated lncRNAs, there were two lncRNAs, ENSG00000235823 (OLMA -LINC, lincRNA) (Fig. 6A) and ENSG00000273001 (RP11-118K6.3, lincRNA) (Fig. 6B), related to AML survival. With the up-regulation of these two lncRNAs, the AML prognosis deteriorated, thus consistent with the poor prognosis of AML when CPT1B was highly expressed.
Characteristics of CN-AML patients by high and low CPT1B expression
Characteristics of CN-AML patients by high and low CPT1B expression
Abbreviations:
Four down-regulated lncRNAs were associated with AML survival. The down-regulated lncRNA ENSG0000 0203804 (ADAMTSL4AS1, processed_transcript) (Fig. 6C) was also called C1orf138. ADAMTSL4 is a member of ADAMTS-like gene family and encodes a protein with seven thrombospondin type 1 repeats. The thrombospondin type 1 repeat domain is present in proteins with diverse biological functions including cellular adhesion, angiogenesis and patterning of the developing nervous system. The down-regulated lncRNA ENSG00000237298 (TTN-AS1, antisense) (Fig. 6D) was found to promote tumor development. The other two down-regulated lncRNAs, ENSG00000267009 (RP11-120M18.2, processed transcript) and ENSG00000269019 (AC005932.1, antisense), have not been reported (Fig. 6F). Low expression of these four lncRNAs predicted worse AML prognosis which was consistent with the poor prognosis of AML patients with high CPT1B expression.
CPT1B expressions were measured via quantitative PCR in BM samples from 324 CN-AML patients in order to verify the prognosis role of CPT1B in AML. The interquartile range of CPT1B transcript level ranged from 1.068 to 5.915. The survival curve (Fig. 7A) plotted using the quartile method enabled the election of a value at 25% as the cut-off for categorising patients into high and low CPT1B expressors. A value of 3.92 (2.37, 7.28) and 0.67 (0.49, 0.89) were the median and interquartile range in high and low CPT1B expressors, respectively. Clinical characteristics of patients with high and low CPT1B expression are shown in Table 1. High expressors had an older age (55 vs 50,
Overexpression of CPT1B is associated with poor clinical outcome in CN-AML patients
In our 324 CN-AML patients, the 3-years overall survival (OS) rate and event-free survival (EFS) rate were 45.5% and 38.9%, respectively. CPT1B in high expression group was significantly associated with short OS (
Multivariate analysis for OS in AML patients (CPT1B)
Multivariate analysis for OS in AML patients (CPT1B)
HR hazard ratio; CI confidence interval;
Survival curves of CN-AML patients. a: Using the quartile method to plot the survival curve of CPT1B in AML. b: High CPT1B expression is associated with poor AML OS. c: High CPT1B expression is related to poor AML EFS.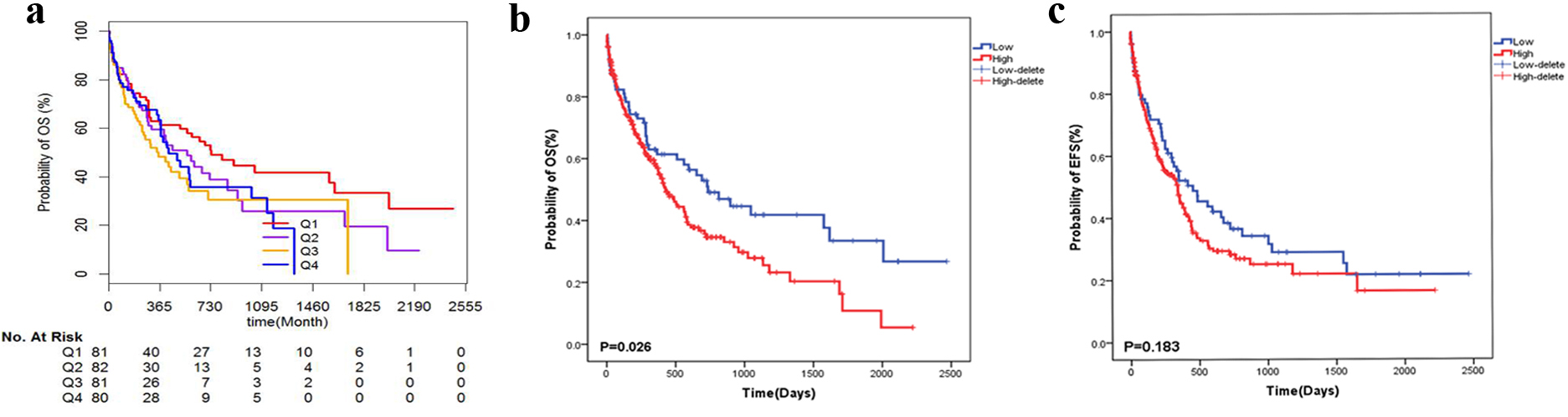
Recent studies have shown that fatty acid oxidation promotes tumorigenesis and development. It can therefore influence the prognosis of cancer patients. For instance, fatty acid rate-limiting enzyme CPT1A has been shown to predict the prognosis of patients. So far, few studies have investigated the role of CPT1B in tumors, especially in CN-AML. In this study, we found that CN-AML patients with high CPT1B expression were older and had high hemoglobin levels. These results supported the hypothesis that increased CPT1B expression is associated with high risk of refractory and relapse in leukemia. Indeed, high CPT1B expressors predicted shorter overall survival in two independent patient cohorts. This study shows that CPT1B is an independent risk factor for poor prognosis of AML.
Expression profiles of CPT1B were analyzed to determine the impact of high CPT1B expression in CN-AML. First, a protein interaction network (PPI) was constructed for 413 significantly up-regulated and 164 significantly down-regulated genes in the high CPT1B expression group using STRING. The PPI network was examined to identify hub genes using the Cytoscape software. The KEGG pathways enrichment analysis revealed many pathways associated with the development of acute myeloid leukemia [21, 22]. The hub genes included in corresponding pathways can be used as potential drug targets. Identification of hallmarks among the ten groups of CN-AML patients via GSEA clarified several upregulated pathways which showed why high CPT1B expression was associated with poor prognosis of CN-AML. Interestingly, the aberrant changes between CPT1B high and low expression groups were related to metabolisms. Fatty acid pathways especially fatty acid metabolism was up-regulated in the high CPT1B expression group (Fig. 2C) This pathway was reported to be involved by CPT1B and to promote tumor cell proliferation. Analysis of mRNA levels further confirmed the prognosis and metabolic function of CPT1B.
By further analyzing the differential miRNAs between the high and low CPT1B expressors, 82 up-regulated miRNAs and 48 down-regulated miRNAs were identified. From the KEGG enrichment analysis combination of the differential miRNAs with the pathways of differential mRNAs, several commonalities were established that pointed out to possible regulatory pathways of CPT1B. miRNAs were then integrated with its pathways to help us find several significant ones:miRNA-18a, which up-regulated in high CPT1B expressor, is highly expressed in many malignancies. The altered miR-18a potentially mediates a regulatory role in a variety of physiological and pathological processes such as cell proliferation, apoptosis, epithelial-mesenchymal transition (EMT), tumorigenesis, cancer invasion and metastasis [23]. Studies had shown that the up-regulated miRNA-182 was associated with poor tumor prognosis [24]. On the other hand, up-regulated miRNA-29a can cause abnormal development of bone marrow and trigger acute myeloid leukemia through abnormal self-renewal ability of hematopoietic progenitor cells [25]. From the properties of miRNAs that were positive correlation with high CPT1B expression, verified the prognostic function of CPT1B from the other side. Furthermore, fatty acid pathways such as fatty acid metabolism, fatty acid elongation, citrate cycle (TCA cycle) revealed a significant change. With these results, it is possible to further reveal the correlation between CPT1B and AML prognosis as well as the metabolic characteristics of CPT1B.
Developing miRNA-mRNA network facilitates understanding the regulatory mechanism between differential miRNAs and mRNAs. The identified miRNA-509 which was upregulated in the high CPT1B expressor was related to the decrease in tumor progression [26]. This indicated the potential of miRNA-509 to mediate cell progress as well as being a progress promoter in AML, which differs with other cancers since AML is aerobic cancer while others are hypoxic cancers. miRNA-339 promotes the development of Stem Cell Leukemia/Lymphoma syndrome via downregulation of the BCL2L11 and BAX pro-apoptotic genes [27]. These results supported the poor prognosis of CPT1B from the related miRNA level. Following the identification of several mRNAs participating in hematopoietic progenitor cell differentiation and metabolic process after function analysis, it is evident that CPT1B high expressor was associated with poor prognosis as well as verifying CPT1B as a metabolic molecule.
To test the hypothesis that lncRNAs mediate the progression of AML due to high CPT1B expression, we analyzed the differential lncRNAs in previous studies. Two lncRNAs, ENSG00000235823 (OLMALINC, lincRNA) and ENSG00000273001 (RP11-118K6.3, lincRNA) were found to be up-regulated. Notably, ENSG00000235823 (OLMALINC, lincRNA) is a long intervening noncoding RNA (lincRNA) found to be involved in the human liver co-expression network (
In summary, this study demonstrates that high CPT1B expression predicts poor survival for patients with CN-AML based on two independent cohorts. A series of mRNAs, miRNAs and lncRNAs that were differentially expressed between high and low CPT1B expression groups were identified via bioinformatics analysis. Enrichment analysis of the differentially expressed mRNAs and miRNAs showed that these RNAs were associated with common pathways such as P53 and mTOR signaling pathways and may form the regulatory network of CPT1B in AML. Analysis of the miRNA-mRNA network revealed several important regulatory relationships. For instance, 2 down-regulated miRNAs and genes co-expressed with CPT1B were found. These biological links can explain the poor prognosis of CPT1B. On the other hand, the luciferase reporting experiment needs to be further developed in the future. A ceRNA network was constructed by combining differentially expressed mRNAs, miRNAs and lncRNAs. This network serves as a reference for further research. Of note, CPT1B was found to be an independent risk factor that predicts poor prognosis and a metabolic molecule that contributes to CN-AML. Moreover, by analyzing the entire possible regulatory network, it further provides ideas for subsequent mechanism research.
Conclusion
In this study, CPT1B was found to be an independent prognostic marker in AML. In addition, a regulatory network of CPT1B in AML was identified. In summary, this study shows the function and mechanisms of CPT1B in AML.
Authors’ contributions
Conception: Qing Ling and Jie Jin.
Interpretation Or Analysis Of Data: Qing Ling and Shihui Mao.
Preparation Of The Manuscript: Qing Ling and Shihui Mao.
Supervision: Jiajia Pan, Wenwen Wei, Yu Qian, Fenglin Li, Shujuan Huang, Wenle Ye, Xiangjie Lin, Jiansong Huang, Jinghan Wang.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 81800199, 81670124, 82070118) and the Natural Science Foundation of Zhejiang Province (LY20H08 0008). The funders had no role in study design, data collection, data analysis, interpretation, writing of this report.
Ethics approval and consent to participate
The study was approved by the Institutional Review Boards of the First Affiliated Hospital of Zhejiang University.
Consent for publication
None of the individual person’s data in this text is for publication.
Availability of data and materials
In this study, 68 CN-AML patients with survival information were obtained from TCGA (
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210043.
sj-pdf-1-cbm-10.3233_CBM-210043.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-210043.pdf
Footnotes
Acknowledgments
We are very thankful to the patients who took part in donating leukemia specimens. We thank all of our laboratory members for helpful discussion.