
Abstract
BACKGROUND:
Gastric cancer (GC) is the third leading cause of cancer worldwide. According to the Lauren classification, gastric adenocarcinoma is divided into two subtypes: diffuse and intestinal. The development of intestinal gastric cancer (IGC) can take years and involves multiple factors.
OBJECTIVE:
To investigate the protein profile of tumor samples from patients with IGC in comparison with adjacent nontumor tissue samples.
METHODS:
We used label-free nano-LC-MS/MS to identify proteins from the tissues samples. The results were analyzed using MetaCore™ software to access functional enrichment information. Protein-protein interactions (PPI) were predicted using STRING analysis. Hub proteins were determined using the Cytoscape plugin, CytoHubba. Survival analysis was performed using KM plotter. We identified 429 differentially expressed proteins whose pathways and processes were related to protein folding, apoptosis, and immune response.
RESULTS:
The PPI network of these proteins showed enrichment modules related to the regulation of cell death, immune system, neutrophil degranulation, metabolism of RNA and chromatin DNA binding. From the PPI network, we identified 20 differentially expressed hub proteins, and assessed the prognostic value of the expression of genes that encode them. Among them, the expression of four hub genes was significantly associated with the overall survival of IGC patients.
CONCLUSIONS:
This study reveals important findings that affect IGC development based on specific biological alterations in IGC patients. Bioinformatics analysis showed that the pathogenesis of IGC patients is complex and involves different interconnected biological processes. These findings may be useful in research on new targets to develop novel therapies to improve the overall survival of patients with IGC.
Introduction
Gastric cancer (GC) is the third leading cause of cancer worldwide, with 1,033,701 new cases and 782,685 related deaths in 2018 [1]. According to the Lauren classification [2], gastric adenocarcinoma is divided into two subtypes: diffuse and intestinal. Intestinal gastric cancer (IGC) is the most frequent subtype in high incidence populations, although its incidence has been declining. IGC is a multifactorial disease that originates from the evolution of sequential lesions. Its development can take years and involves lifestyle, aging, genetics, socioeconomic factors, and infectious agents such as Helicobacter pylori (HP) [3, 4, 5]. HP infection can elicit continuous inflammation, immune response and abnormal epithelial cell proliferation, inducing a metaplastic process that can lead to atypia and occasional neoplasia characterizing the development of IGC [6, 7]. Despite the advances in diagnosis, GC is often detected in late stages due to the nonspecific symptoms. The five-year survival rate is only 20%, and chemotherapy and surgery have limited value in the treatment of advanced cases. Adding to this problem, there is a lack of targeted therapies that use molecular markers [3]. To understand the carcinogenesis of GC, molecular high-throughput studies, including genomics, transcriptomics and proteomics, have been performed. In cancer, these studies mainly address the quantitative and qualitative differences in the protein alterations in tumor and nontumor tissues since these alterations are involved in the carcinogenic process [8]. The use of these technologies for studying GC enables the identification of proteins in different biological specimens, such as blood, gastric fluids, tumor tissues and tumor cells [9]. However, the use of proteomic approaches to study IGC has not been extensively explored compared to their application to other solid tumors. Studies focusing on signaling pathways and interaction networks in IGC are indispensable since the regulatory control of protein-protein interactions (PPIs) plays a critical role in biological pathways in cancer, including apoptosis, cell proliferation, trafficking and angiogenesis. Thus, alterations in proteins and PPIs are related to malignancies and constitute critical targets for treating cancer [10]. A comprehensive analysis of proteomic alterations and their interactions using bioinformatics approaches would greatly benefit our understanding of the molecular background of IGC. Thus, with the aim of further understanding the proteomic changes in the occurrence and development of IGC, we used high-throughput proteomic analysis to investigate the protein profiles of tumor samples from IGC patients and compared them to adjacent nontumor tissue samples from the same patient using label-free nano LC-MS/MS. We identified 429 differentially expressed proteins whose pathways and processes were related mainly to protein folding, apoptosis, and immune response. The PPI network of these altered proteins showed enrichment modules related to the regulation of cell death, the immune system, neutrophil degranulation, the metabolism of RNA and chromatin DNA binding. We also identified 20 key hub proteins, and we assessed the prognostic value of the expression of the genes that encode them. Among them, the expression levels of four hub genes were significantly associated with the overall survival of IGC patients. Protein alterations and their relations to the specific signaling pathways found in this study will help us to better understand the tumor biology of IGC patients, highlighting new potential targets for further investigation.
Material and methods
Patients and study design
The study was conducted in accordance with the Declaration of Helsinki, and all participants signed an informed consent form approved by the National and Institutional Ethics Committee. Tumor samples (from biopsy) were obtained from patients (
Study design. The study schema is illustrated with the methods used and patient populations. Tumor tissues and nontumor adjacent tissues controls from IGC patients were submitted to protein extraction followed by label free proteomic approach. Chromatography coupled to mass spectrometry was used for protein identification and quantification. After statistical analysis and data mining, a list containing a profile of upregulated and downregulated proteins from IGC patients were generated. The list of proteins was submitted to functional enrichment analyses using important bioinformatics tools to identify significant biological pathways and processes. Subsequently, the interaction between the proteins was determined and the top interacting proteins were selected. The genes of the final selected proteins were then evaluated regarding its prognostic value in a cohort of 201 IGC patients.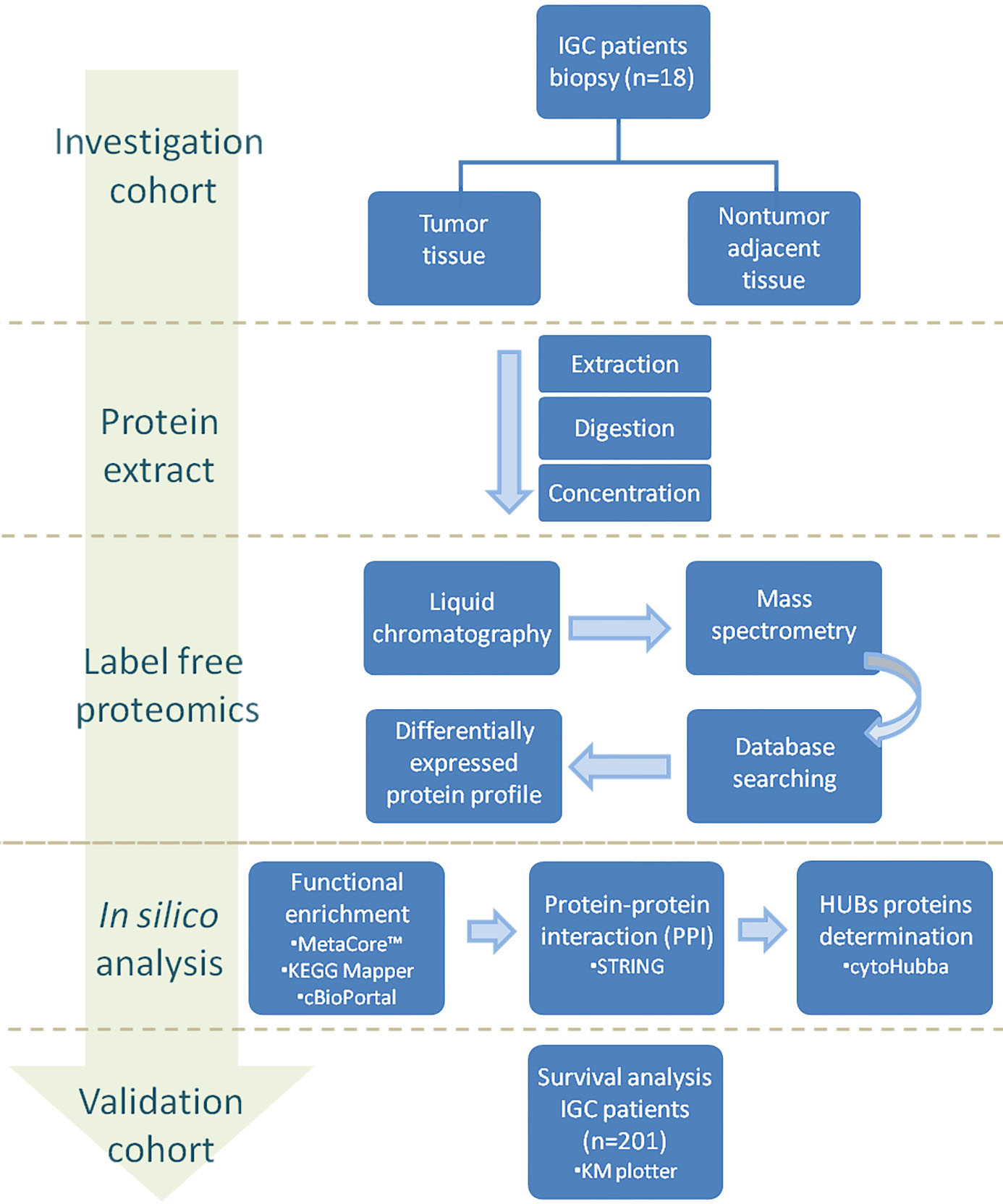
During endoscopic evaluation, biopsies from patients were obtained. From the same stomach area, half of the biopsies were prepared for histopathologic evaluation, while the other half were frozen and sent to the National Tumor Biobank at INCA, where they were stored at
In the histopathologic examination, H. pylori status was investigated initially with Wayson stain and analyzed by two independent experienced pathologists. At that time, the first status of H. pylori was defined. To further validate these results, immunohistochemistry was performed using the rabbit monoclonal anti-Helicobacter pylori (SP48) antibody (Roche Ltd, Basel, Switzerland).
Protein extraction, concentration and digestion
Total protein was extracted from tumor and adjacent nontumor tissues, and the samples were concentrated as previously reported [11] and quantified using the Braford method [12]. Two hundred micrograms of protein was used for tryptic digestion, as previously reported [11]. After digestion, 5 mM ammonium formate with 5% acetonitrile was added to the sample for a final volume of 160
Liquid chromatography and mass spectrometry
The samples were subjected to nanoscale chromatographic separation (2DnanoLC) using the nanoACQUITY UPLC system from Waters
Identification, quantification and in silico analysis
Database searching and protein quantification were performed as previously reported [13]. Briefly, ProteinLynx Global Server v.3.0 (PLGS) with ExpressionE software was used to process the spectra and databank search results. The UniProtKB databank (release 2017_07) was used with manually reviewed annotations. Only the proteins found in all replicates under each condition were subjected to analysis with the PLGS Expression
Survival analysis
The survival analysis of genes was performed using KM plotter (
Results
Proteomic profile evaluation of the IGC tumor samples
To identify proteomic alterations, we used label-free protein quantification by mass spectrometry to compare the proteomic profiles of tumor and adjacent nontumor tissue samples (from biopsy) from Brazilian patients (
Functional enrichment pathways related to the 429 differentially expressed proteins in IGC patients
Functional enrichment pathways related to the 429 differentially expressed proteins in IGC patients
Functional enrichment process networks related to the 429 differentially expressed proteins in IGC patients
PPI analysis of 429 differentially expressed proteins exclusively found in IGC patients. Network nodes represent interacting proteins. Nodes also indicate the three dimensional protein structures that are known or predicted. Red nodes represent the proteins related to the regulation of cell death. Green nodes represent the proteins related to the immune system. Blue nodes represent the proteins related to neutrophil degranulation. Pink nodes represent the proteins related to the metabolism of RNA. Yellow nodes represent proteins related to chromatin DNA binding. Different line colors indicate the types of evidence supporting these proposed associations. Proteins not connected with the network were removed for better visualization.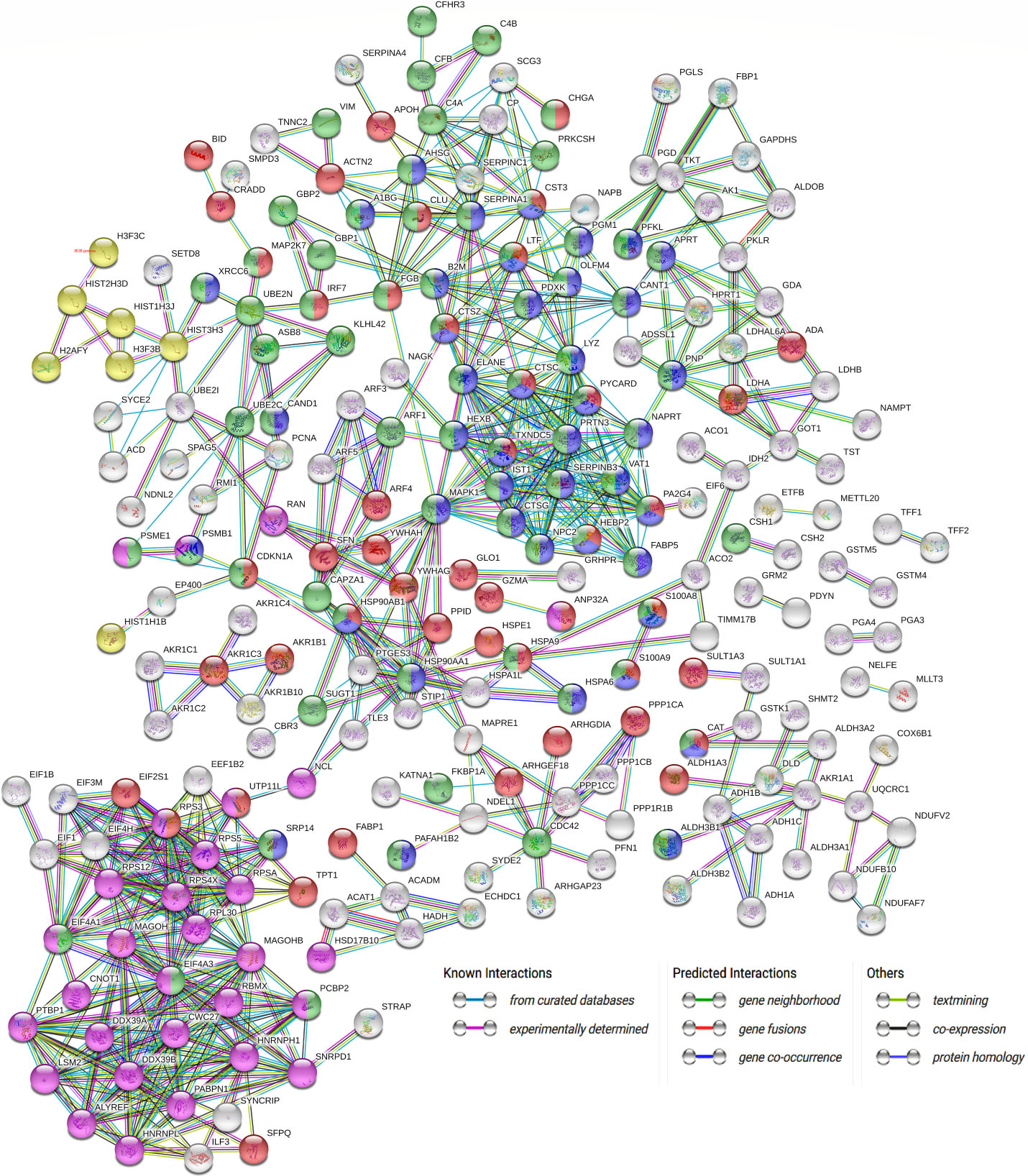
Top 20 hub proteins in the PPI network, ranked by the maximal clique centrality method
Based on proteomic results, we used MetaCore™ software (GeneGo Inc., Encinitas, USA) to investigate the pathways and biological processes related to the 429 differentially expressed proteins. The results showed that enriched pathways and processes were related mainly to protein folding, apoptosis, and immune response (Tables 1 and 2). The top 10 signaling pathways and process network results showed that a group of molecular chaperones (upregulated HSP70, HSP90 alpha, HSP90, HSP90 beta, and HSP10), an important family of proteins commonly expressed in response to stress conditions and also known as heat shock proteins (HSPs), was overrepresented in these pathways (Supplementary S3). HSPs were found in chaperone-specific pathways and in networks related to gastric cancer and apoptosis (Table 1). We also found proteins related to apoptosis pathways in these patients. A protein structurally similar to caspase-8, CASP8 and FADD-like apoptosis regulator (c-FLIP, which was downregulated) that functions as a crucial link between cellular survival and death was altered, and BH3-interacting domain death agonist (BID, which was upregulated), a member of the BCL-2 protein family, was also altered (Supplementary S4). We also identified the downregulation of important proteins related to the differentiation of gastric mucosa and its protection, such as trefoil factor 1 and 2 (TFF1 and TFF2), chromogranin A, regenerating family member 1 alpha (ICRF, also known as REG1A) and the mitogen factor gastrin 17 (Supplementary S5). Moreover, we observed that histones, important components in gene regulation, had deregulated expression (histones from H1 and H3 families were upregulated, and the H2 histone was downregulated) and an N-lysine methyltransferase was upregulated. Furthermore, enrichment analysis of the top 10 process networks showed downregulated regulatory proteins, such as the 14-3-3 family proteins (14-3-3 sigma, 14-3-3 gamma, 14-3-3 theta, and 14-3-3 eta) and mitogen-activated protein kinase 1 (MAPK1), in networks related to inflammation, cytoskeleton regulation and apoptosis.
Protein-protein interaction (PPI) in IGC patients
To investigate the interactions among the 429 differentially expressed proteins in IGC patients, we built a PPI network. A complex protein-protein association was observed in which most proteins had direct or indirect interactions through the number of observed nodes (Fig. 2). Corroborating our previous results, the following biological processes were overrepresented: the regulation of cell death (62 proteins), the immune system (63 proteins), and neutrophil degranulation (45 proteins). Furthermore, the metabolism of RNA (30 proteins) and chromatin DNA binding (7 proteins). Some proteins in the PPI network had a high number of interactions, such as MAPK1, which showed 27 interactions and was connected to proteins involved in the regulation of cell death, the immune system and neutrophil degranulation processes. Elastase neutrophil expressed (ELANE) is a serine protease that hydrolyzes proteins within specialized neutrophil lysosomes and has 22 interactions. Proliferation-associated 2G4 (PA2G4), a protein related to growth regulation and differentiation in cancer, showed 20 interactions. Moreover, proteins from a family of heterogeneous nuclear ribonucleoproteins, polypyrimidine tract-binding protein (PTBP1), displayed 18 interactions and heterogeneous nuclear ribonucleoprotein K (HNRNPK), with 18 interactions, are all involved in processing mRNAs and act as trans-factors in regulating gene expression. DExD-box helicase 39B (DDX39B), with 16 interactions, is involved in various cellular processes, such as mRNA export, splicing and translation.
Analysis of hub differentially expressed genes in IGC patients. (a) PPI network of hub protein nodes representing highly interacting proteins. Nodes also indicate the three dimensional protein structures that are known or predicted. Blue nodes represent the proteins related to neutrophil degranulation. (b) Enrichment analyses of hub proteins. Top ten significantly ranked enriched biological processes, reactome pathways, molecular functions and cellular components of hub proteins. (c) The oncoPrint plot showing the different genomic alterations in a set of 109 IGC patient samples. (d) An overview summarizing the alterations in the 20 hub genes in the genomics datasets of IGC in the TCGA database.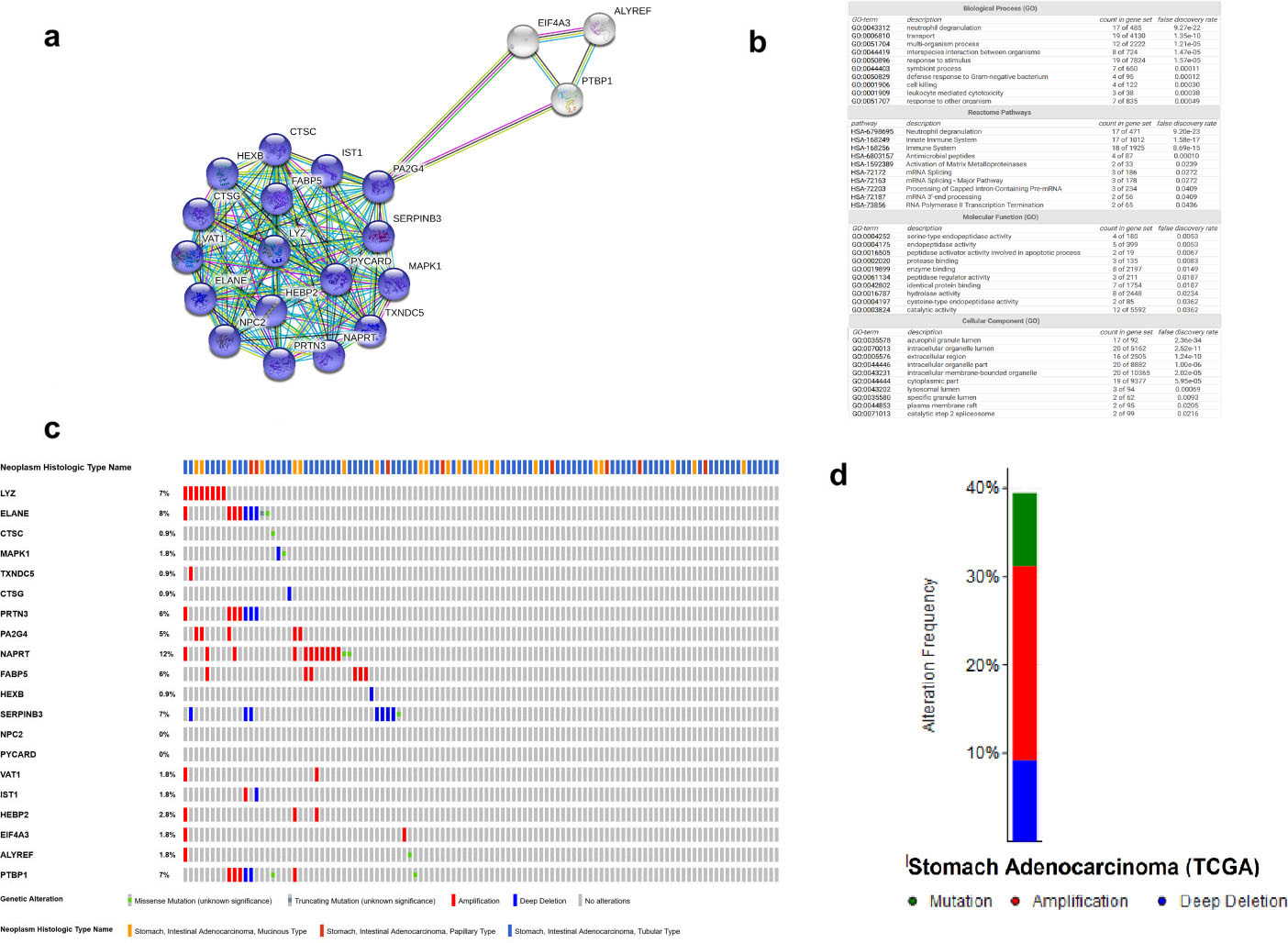
The cumulative survival curves of the hub genes in IGC patients plotted using KM plotter. (a) IST1 (also known as KIAA0174). (b) VAT1. (c) ALYREF. (d) HEXB. 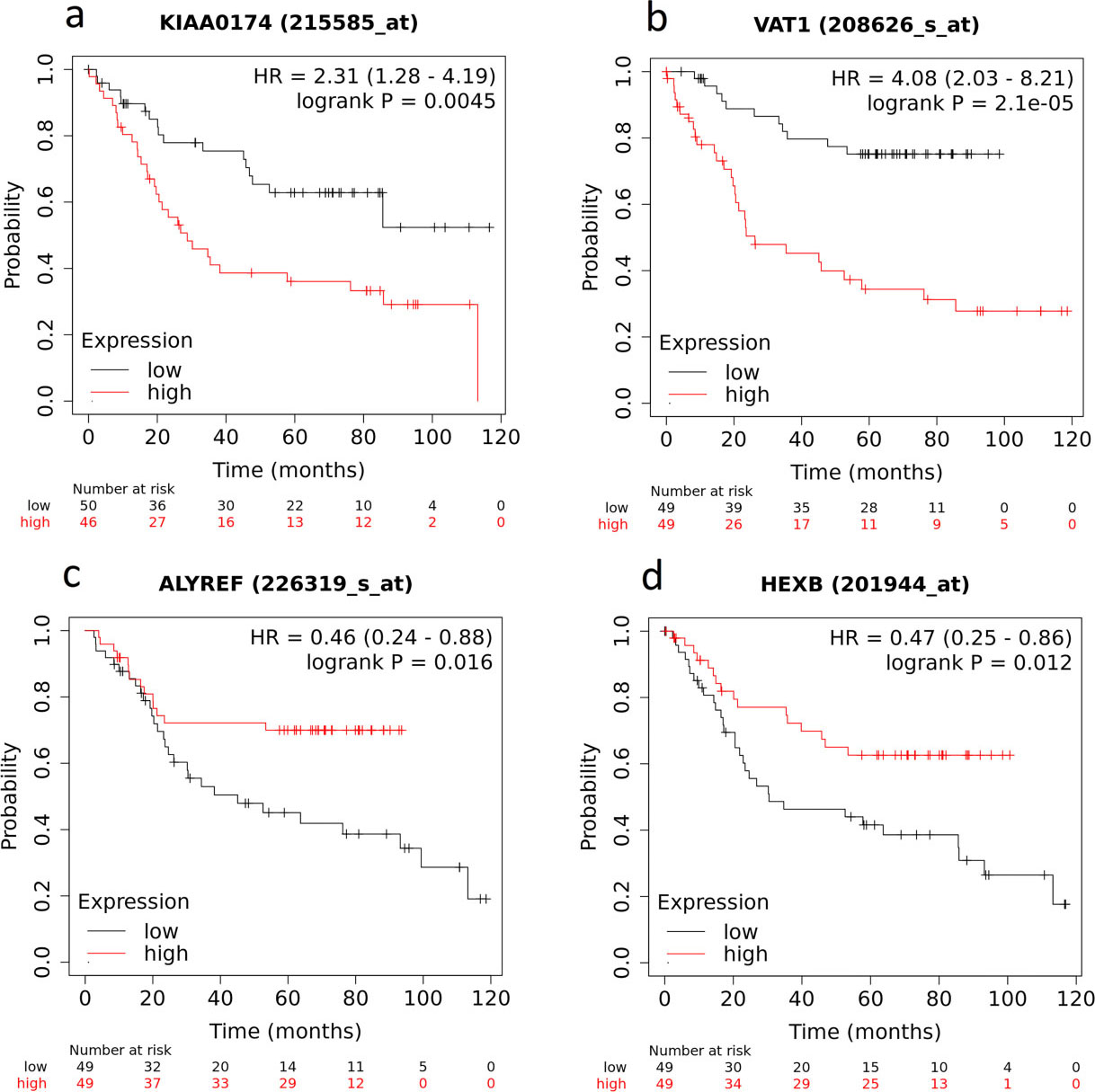
Hub proteins are characterized by a large number of interactions and are frequently essential in multiple pathways; therefore, they are important for disease development. Thus, to identify highly connected protein nodes, we determined the hub proteins from the PPI network based on the STRING interaction data. We used the plug-in “cytoHubba” to rank the nodes by their network features through topological analysis methods. The hub proteins were chosen based on the following standards: the top 20 nodes from the network identified by the maximal clique centrality (MCC) analysis method, which has better performance regarding the precision of predicting essential proteins [16]. The results indicated 20 proteins that were identified as hub proteins in the PPI network. Ten of these hub proteins were upregulated, and 10 were downregulated (Table 3). Furthermore, the results of the enrichment analysis of hub proteins in the STRING interaction data showed a significant representation of neutrophil degranulation and immune system biological processes and pathways, as well as intracellular organelles for cellular components (Fig. 3a and b). Figure 3c and d shows the genomic alterations of the 20 hub genes according to cBioportal (data from the TCGA, Firehose Legacy). The 20 hub genes were altered in 43 (39%) of the 109 sequenced patients, mutations 8.26% (9 patients), deep deletions 9.17% (10 patients) and amplification 22.02% (24 patients).
Survival analysis of the hub genes in KM plotter
Due to the small size of our cohort and the lack of follow-up of the patients, it was not possible to evaluate the prognostic value of hub proteins. To address this question, we decided to analyze the prognostic value of the genes that encode the 20 hub proteins in public software (KM plotter) with data sourced from 1,440 gastric cancer patients [18, 19]. After filtration of these gastric cancer samples, data from 201 IGC patients were used in survival analysis. Of the 20 hub genes, four were significantly associated with overall survival (OS). High expression of IST1 (also known as KIAA0174) (log-rank
Discussion
The purpose of this study was to identify global proteomic alterations in a Brazilian cohort of patients with ICG. We used label-free nano-LC-MS/MS and in silico approaches to assess the relevant biological alterations in tumors from IGC patients, focusing on alterations in biological pathways and PPI networks that are not available in the current literature.
Overall, when we compared IGC tissue samples with their respective adjacent nontumor samples, we observed that biological pathways and processes inherent to cellular and systemic homeostasis, such as protein folding, cytoskeleton rearrangement, apoptosis and the immune response, were commonly altered. To that extent, we observed that some proteins, especially those related to protein folding, such as HSP, were commonly upregulated and related to chaperone activity, apoptosis and the immune system.
HSPs have a cytoprotective function that is important to cells under normal conditions and constitute the first defense responses to stress conditions [20, 21]. The main activity of HSPs is their chaperone function, through which they are important to the proper folding of proteins, maintaining their structures and functions in cells even under adverse conditions [22, 23]. In cancer cells with high metabolic requirements, it is common to observe the deregulation of these proteins, since the majority of relevant oncoproteins require high levels of HSP chaperones to ensure proper protein folding, stabilization, aggregation, activation, operation and degradation. Among the HSPs, upregulated members from the HSP90 family were found; these proteins are the most studied HSPs, with altered expression in several solid tumors [24]. In gastric cancer, high expression of HSP90 is associated with the staging, proliferation and aggressiveness of the tumor. We also observed dysregulation of HSP70 family proteins (upregulation of HSPA1L and downregulation of HSPA6), which are related not only to specific antitumor immunity but also to regulatory T cell responses [25]. HSPA1L is known to increase cell survival and protection against apoptotic and necrotic stimuli [26]. In addition, it is related to the progression of several solid tumors and to poor prognosis for patients [27, 28]. High expression of HSP70 was observed in GC cell lines [29]. HSP70 overexpression also correlated with poor prognosis in patients with esophageal squamous cell carcinoma and potentially with the progression of other tumors, such as cervical cancer [30], breast cancer [31] and nasopharyngeal carcinoma [32]. The 10 kDa mitochondrial heat shock protein HSPE1, an HSP10 family protein, was also upregulated. This protein is related to immunomodulation, differentiation and proliferation, and it can inhibit apoptosis [33, 34]. High levels of this protein were detected in several tumors [34, 35, 36, 37, 38]. From all these observations, it is possible that pathways related to apoptosis and survival are highly impacted in patients with ICG through the dysregulation of HSPs. Among the identified proteins, the upregulation of BID was found. BID is a pro-apoptotic protein member of the BCL-2 family and is an important regulator that links components of the extrinsic and mitochondrial (intrinsic) pathways of apoptosis [39].
We noted that differentiation of the gastric mucosa pathway was enriched through alterations in important proteins related to its normal function, such as the downregulation of CHGA, TFF1, TFF2, and ICRF (also known as REG1A). In a previous work by our group [40], we verified the downregulation of mRNA levels of CHGA, TFF2 and ICRF (also known as REG1A) in a common molecular signature for intestinal gastric cancer worldwide, corroborating these findings. The genes of the TFF family play important roles in the digestive system of mammals, are mainly related to epithelial cell reconstruction, protection of gastric mucosa, signal transduction and regulation of apoptosis and proliferation, and may act in tumor suppression or promotion. TFF1 and TFF2 are downregulated in gastric cancer tissue compared with normal tissue, and TFF2 downregulation may be associated with hypermethylation [41, 42].
Proteins need to interact with each other to fulfill their functions, and this characteristic is fundamental in most biological processes. Thus, to better comprehend the biological function is essential to understand protein interactions. Highly connected proteins (hubs) in PPIs are often essential to network architecture and functioning [43]. Since high-throughput proteomic analysis has acquired prominence in cancer research, drug targeting investigations using hub proteins and their interactions could be especially interesting [44, 45, 46].
PPI analyses showed the enrichment of proteins with direct and indirect interactions in processes related to apoptosis and neutrophil degranulation, and according to STRING enrichment analyses, the hub proteins are highly involved in neutrophil degranulation. Mature neutrophils represent approximately 50–70% of all leucocytes in adult peripheral blood and are important components of the tumor microenvironment [47]. Neutrophil functionality depends on its cytoplasmic granule composition, which includes important proteins that can promote carcinogenesis [48]. One of these proteins, neutrophil elastase (ELANE), which was also identified as a hub protein, was upregulated. The protumorigenic role of ELANE has been observed in lung, breast, and colon cancers [49, 50, 51]. Notably, in the PPI analysis results, ELANE interacted with other important hub proteins, including the upregulated MAPK1. MAPK1 was the top interacting protein in the PPI analysis, and fourth in the hub analysis; therefore, MAPK1 may be a master regulator related to both apoptosis and neutrophil degranulation processes in IGC patients.
It is interesting to note that in our cohort, all patients were positive for Helicobacter pylori (HP) infection. In addition, an important protein related to tight junctions, occludin, was downregulated in these patients. Long-term infection with HP leads to the disruption of gastric epithelial junctional structures through the downregulation of proteins related to membrane integrity, such as E-cadherin and occludin, and contributes to tissue alterations related to carcinogenic processes [52, 53]. PPI analyses also revealed clusters of interacting proteins related to chromatin DNA binding, where histone variants were dysregulated. The accessibility of DNA for transcription is regulated by complex posttranslational modifications of those histones, such as acetylation, methylation, phosphorylation, ubiquitination, and ADP-ribosylation [54, 55]. MCF-7 cells from breast cancer treated with estrogenic compounds showed an increase in the proliferative rates accompanied by changes in the levels of histones [56]. Additionally, increased histone variant expression is a strong prognostic biomarker in cervical cancer patients [57]. Based on this information, it is reasonable to affirm that alterations in histone variant expression can regulate gene expression and are related to the malignancy of tumors.
In CG, HP infection was observed to induce histone modifications, such as the dephosphorylation of histone H3, thereby regulating gene expression, the cell cycle and cell proliferation [58, 59], and acetylation of H4, which is associated with p21 gene expression in gastric epithelial cells [60]. However, there is little information about the impact of HP infection on the levels of histone expression or the consequences of any such impact in CG patients. We also observed a cluster of proteins related to mRNA metabolic processes, especially proteins from the heterogeneous nuclear ribonucleoprotein (hnRNP) family, such as upregulated SYNCRIP and downregulated HNRNPL. These proteins are RNA-binding proteins that control posttranscriptional regulation, mediating alternative splicing and polyadenylation, thereby directly influencing protein abundance and diversity. SYNCRIP overexpression is related to cell proliferation, growth and survival and maintains the undifferentiated status of leukemic cells [61]. However, there is no information on this protein in GC patients. In contrast to our results, HNRNPL was found to be upregulated in GC patients from a Chinese population; however, the HP status of these patients was not available [62]. These results indicate that HP infection may have an impact on gene expression through histone variants and hnRNP protein alterations.
Furthermore, among the 20 hub proteins identified, IST1 (also known as KIAA0174), VAT1, ALYREF, and HEXB could predict the overall survival. IST1 regulates the disassembly of endosomal sorting complexes in yeast and is related to poor prognosis in GC [63]. Vesicle amine transport 1 (VAT1) is upregulated in glioblastomas and promote migration [64]; however, there is no information about this protein in GC. ALYREF is a nuclear protein that acts as a chaperone, thereby regulating DNA binding, dimerization, transcriptional activity, and mRNA export [65, 66], and is significantly related to the survival outcome in hepatocellular carcinoma [67]. Beta-hexosaminidase subunit beta (HEXB) is a lysosomal enzyme involved in the hydrolysis of ganglioside GM2 to GM3, and its protein expression is related to poor survival in malignant melanoma [68].
This study reveals important findings to comprehend IGC development; however, it has some limitations. The work had initially a small set of the patients with IGC (
In conclusion, based on the differences in the expression of the proteins observed in the proteomic profile of IGC, we were able to identify important proteins and biological processes by integrating comprehensive proteomic approaches and bioinformatics analysis, which showed that the pathogenesis of IGC patients is complex and involves different interconnected biological processes. The present study revealed a considerable number of differentially expressed proteins, which are mainly involved in protein metabolism, cytoskeleton rearrangement, apoptosis, immune response RNA metabolism and chromatin DNA binding. We also identified 20 hub proteins that may have important roles in the molecular background of IGC development. Moreover, the overall survival prediction of IST1 (also known as KIAA0174), VAT1, ALYREF, and HEXB may be useful in researching new targets to develop novel therapies and individualized treatments to improve the overall survival of patients with IGC.
Authorship contributions
Conception: E.C.S., R.B.
Interpretation or analysis of data: E.C.S., R.B., P.V.F., P.V.F.
Preparation of the manuscript: E.C.S., R.B.
Revision for important intellectual content: E.A.
Supervision: E.A.
Supplementary data
The supplementary files are available to download from
sj-pdf-1-cbm-10.3233_CBM-203225.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-203225.pdf
sj-docx-1-cbm-10.3233_CBM-203225.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-203225.docx
sj-xls-1-cbm-10.3233_CBM-203225.xls - Supplemental material
Supplemental material, sj-xls-1-cbm-10.3233_CBM-203225.xls
Footnotes
Acknowledgments
This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Ministério da Saúde (MS), and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). We would like to thank the National Tumor Biobank (BNC) at INCA for providing the tissue samples.
Conflict of interest
The authors declared that they have no competing interests.