
Abstract
BACKGROUND:
Colorectal carcinoma (CRC) is one of the most leading cause of cancer death all over the world. The tumor immune microenvironment is illustrated to be necessary for the progress of CRC. And the accumulating evidence indicated that tumor mutation burden (TMB) is effective in differentiating responding population of immune checkpoint inhibitor (ICI) therapies in various cancers. In this study, we aimed to evaluated the potential relationship between TMB and the recurrence risk of CRC.
METHODS:
The transcriptomic and clinical data of CRC patients were collected from The Cancer Genome Atlas (TCGA) database (
RESULTS:
Firstly, we accessed the distribution of TMB and preferences at the gene and mutation level using somatic mutation data from TCGA data about CRC. We identified that high TMB predicted better prognosis of CRC patients. Secondly, the differentially expressed genes (DEGs) between the low TMB and high TMB group was clarified. Then the protein-protein interaction (PPI) analysis was performed, and the results confirmed ten hub genes among the DEGs. Utilizing the GEPIA web-tool, we discovered that GNG4 was up-regulated in tumor tissues, and GNG4 was related to the overall survival (OS) and tumor free survival (TFS) of CRC patients. Therefore, we considered GNG4 was essential for the tumor immune microenvironment of CRC. Furthermore, we also accessed the protein level of GNG4 in CRC and liver metastases from CRC.
CONCLUSIONS:
In this study, GNG4 was demonstrated to be the key element of the CRC TMB, which will be essential for the ICI therapy of CRC. Besides, GNG4 was up-regulated in CRC and liver metastases from CRC tissues. Thus, we thought that GNG4 might play an important role in colorectal cancer TMB and induce its metastasis in liver.
Introduction
The recent studies presented that colorectal cancer is a major cause of incidence and mortality worldwide, and the incidence rate of CRC is increasing in the several decades [1]. Thus, it has been considered as a type of malignant that poses a great threat to human survival, especially in the developing countries [1, 2]. The wide epidemiological and observational evidence that the risk of colorectal cancer is strictly related to obesity, aging, alcohol abuse, and chronic intestinal inflammation [3, 4]. In addition, the dietary fiber is reported to reduce the risk of CRC [4, 5]. Over the years, researchers have treated the carcinogenesis of CRC as a stepwise process, beginning from single mutational event in a cell, until detectable malignancy. At the same time, aiming to block the course of this process, or almost thought to be tested in several models [1, 6, 7]. Due to the genetic mutations and epigenetic modifications in CRC, the molecular classification of CRC provides the basis for evaluation of prognostic, and predictive markers.
In order to elevate the most likely benefit from immune checkpoint blockade (ICB), reliable biomarkers were detected by many studies. Programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1) were the biomarkers approved for selecting patients for response to ICB [8, 9]. Genetic aberrant variants paly the important part in the pathogenesis of various tumors, including missense mutations (point mutations that change the amino acid codon), synonymous mutations (silent mutations that do not alter amino acid coding), insertions or deletions, and copy number gains and losses [10]. Due to these genomic changes in tumor, the concept of tumor mutation burden (TMB) was revealed. In multiple cancers, TMB is reported to be the most robust, effective and clinical verifiable biomarker. In the neo-antigen peptides treated none small-cell lung cancer (NSCLC) patient cohort, TMB value is highly associated with tumor immune response. In addition, the results of clinical trial CheckMate-032 study showed that high TMB level group was more sensitive to the Nivolumab single agent treatment and Nivolumab plus Ipilimumab combination therapy [11, 12, 13]. However, the TMB in CRC patients remains unknown.
Utilizing somatic mutation data from TCGA data about CRC, we accessed the distribution of TMB and preferences at the gene and mutation level. Then we discovered that high TMB value was correlated to the good prognosis of CRC patients. Besides, the patients were divided into high TMB cohort and low TMB cohort. Comparing these two groups, DEGs were clarified. Then the protein-protein interaction (PPI) analysis was carried out, and the results confirmed ten hub genes among the DEGs. Subsequently, we observed that GNG4 was up-regulated in tumor tissues, and GNG4 was related to the overall survival (OS) and tumor free survival (TFS) of CRC patients. Therefore, we considered GNG4 was essential for the tumor immune microenvironment of CRC.
Methods and materials
TMB calculation
TMB was defined as the number of somatic mutations in the coding region per megabase. In this study, we calculated the TMB through the following formula: TMB
Data processing of DEGs
In order to find out the DEGS between CRC and normal samples, we used the GEO2R online analysis tool in NCBI (
GO and KEGG pathway analysis of DEGs
Gene Ontology (GO) is used widely in functional annotation and enrichment analysis, which including three components: biological process (BP), molecular 105 function (MF), and cellular component (CC). KEGG is a database resource for collecting large number of data about molecular level information, biological pathways, chemical substances which is generated by high-throughput experimental technologies. With the help of web-tool called Database for Annotation, Visualization and Integrated Discovery (DAVID) (
PPI network construction and hub gene identification
The DEGs were uploaded to the Search Tool for the Retrieval of Interacting Genes (STRING) database analysis platform to obtain a PPI map. The PPI pairs which possessed a combined score
Immunohistochemistry assay?
The paraffin-embedded CRC and adjacent normal tissue from our center were used for immunohistochemistry. The tissues were cut at 5 mm, deparaffinized in xylene, and rehydrated in a series of graded alcohol dilutions. Heat epitope retrieval was done for 20 min in a target-retrieval solution in the condition of pH 7.5. The histological sections were incubated with a rabbit polyclonal anti-GNG4 antibody at the dilution of 1:500 overnight at 4
Statistical analysis
All statistical analyses were performed using SPSS 22.0 (SPSS Inc., Chicago, IL, USA) and R software, version 3.4.3 (The R Foundation for Statistical Computing,
Result
Distribution of TMB in CRC based on TCGA
About 382 CRC patients collected from TCGA database were involved in this study. Then the WES data of CRC were analyzed. The CRC TMB levels were evaluated through the calculating formula: TMB
The distribution of TMB in CRC based on TCGA database. A. The variant classification (left panel), variant type (middle panel), and SNV class (right panel) of mutated genes involved in CRC tumors. B. The TOP 10 mutated genes and variant classification of CRC tissues. C. The water-fall diagram indicated the TOP 35 mutated genes and their variant types in CRC tissues based on TCGA database.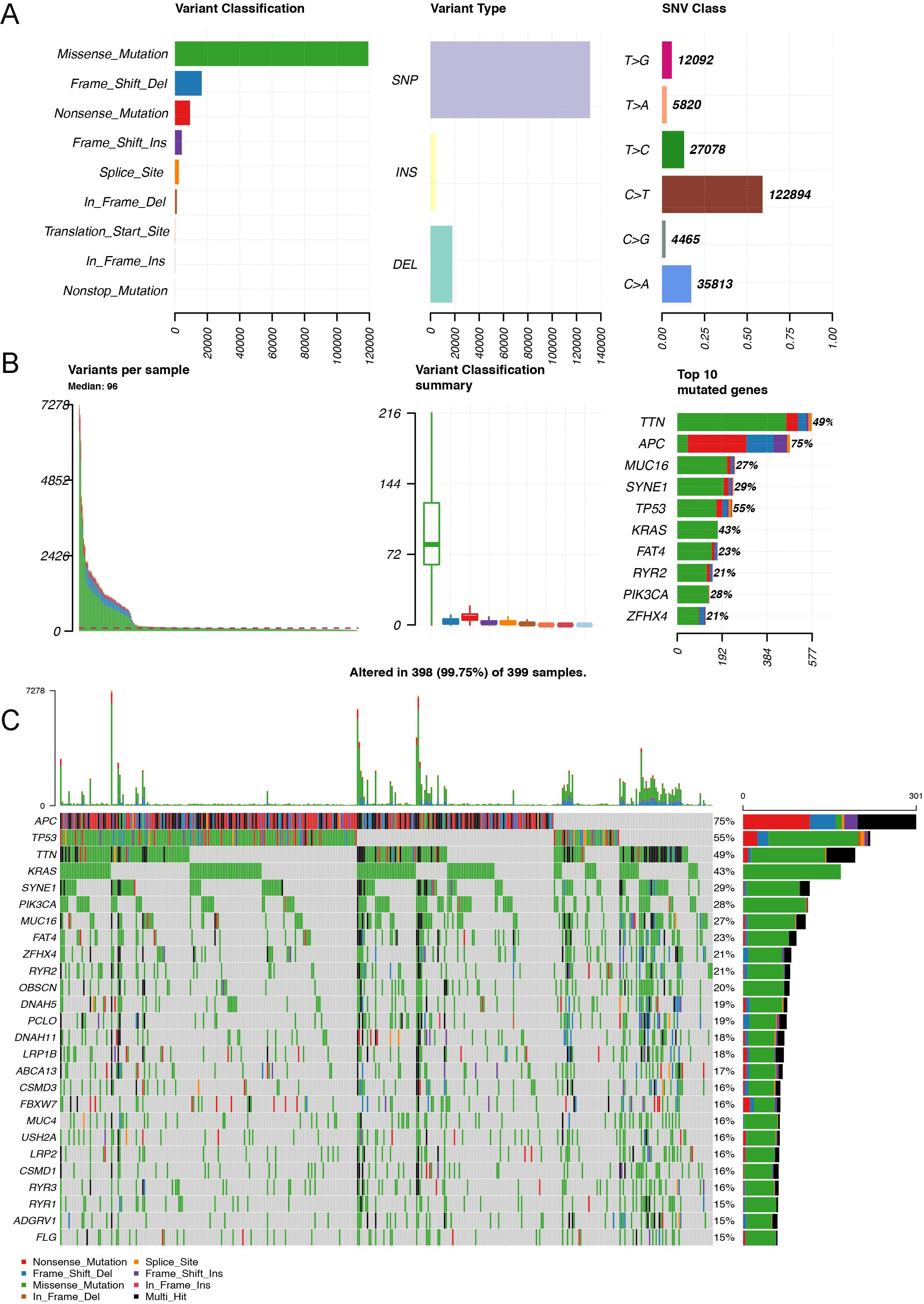
The associations of clinical features and TMB in CRC. A. The co-occurrence of mutated genes in CRC. B. Higher TMB predicted better prognosis in CRC patients. C. The CRC tissues in Stage I and Stage II showed increased as compared to Stage III and Stage IV. In this part, we used the false discovery rate (FDR) to adjust the 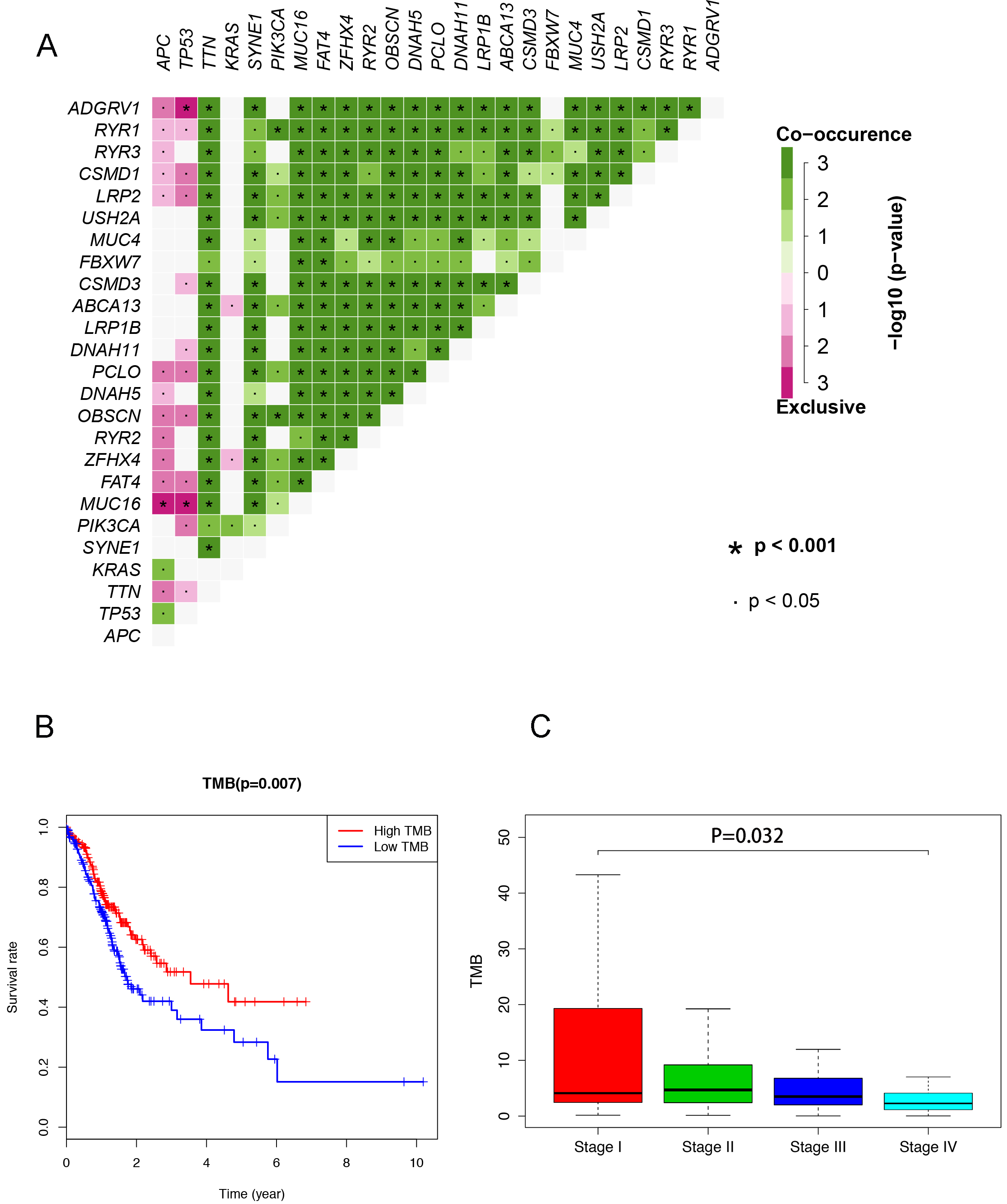
Many studies have demonstrated that the cancer-causing genes could be co-occurring or show strong exclusiveness in their mutation pattern with high TMB. Here, the top genes set ADGRV1, RYR1, CSMD1, LRP2, USH2A, MUC4, FBXW7, CSMD3, ABCA13, LRP1B showed a strong co-occurrence and gene set APC, TP52, TTN, KRAS, SYNE1, PIK3CA, MUC16, FAT4, ZFHX4, and RYR2 (Fig. 2A). In addition, as a CBI treatment selection biomarker, TMB is reported to be associated with the prognosis of various malignancies. Our findings observed that the overall survival of CRC patients became better followed by the higher level of TMB (Fig. 2B). Then the integrative analysis between the clinical characteristics and TMB was carried out in this research. As shown in Fig. 2C, the TMB in stage I and stage II was higher as compared to the TMB in stage III and stage IV of CRC.
Identification of TMB related DEGs in CRC
In order to screen out the TMB related differential expressed genes (DEGs) about CRC, we divided the tumor tissues into low TMB cohort/high TMB cohort. According to the criteria of
The GO and KEGG enrichment analysis of TMB related DEGs in CRC. A. The GO analysis results of TMB related DEGs. B. The KEGG enrichment analysis results of TMB related DEGs. C. The establishment of PPI network based on TMB related DEGs.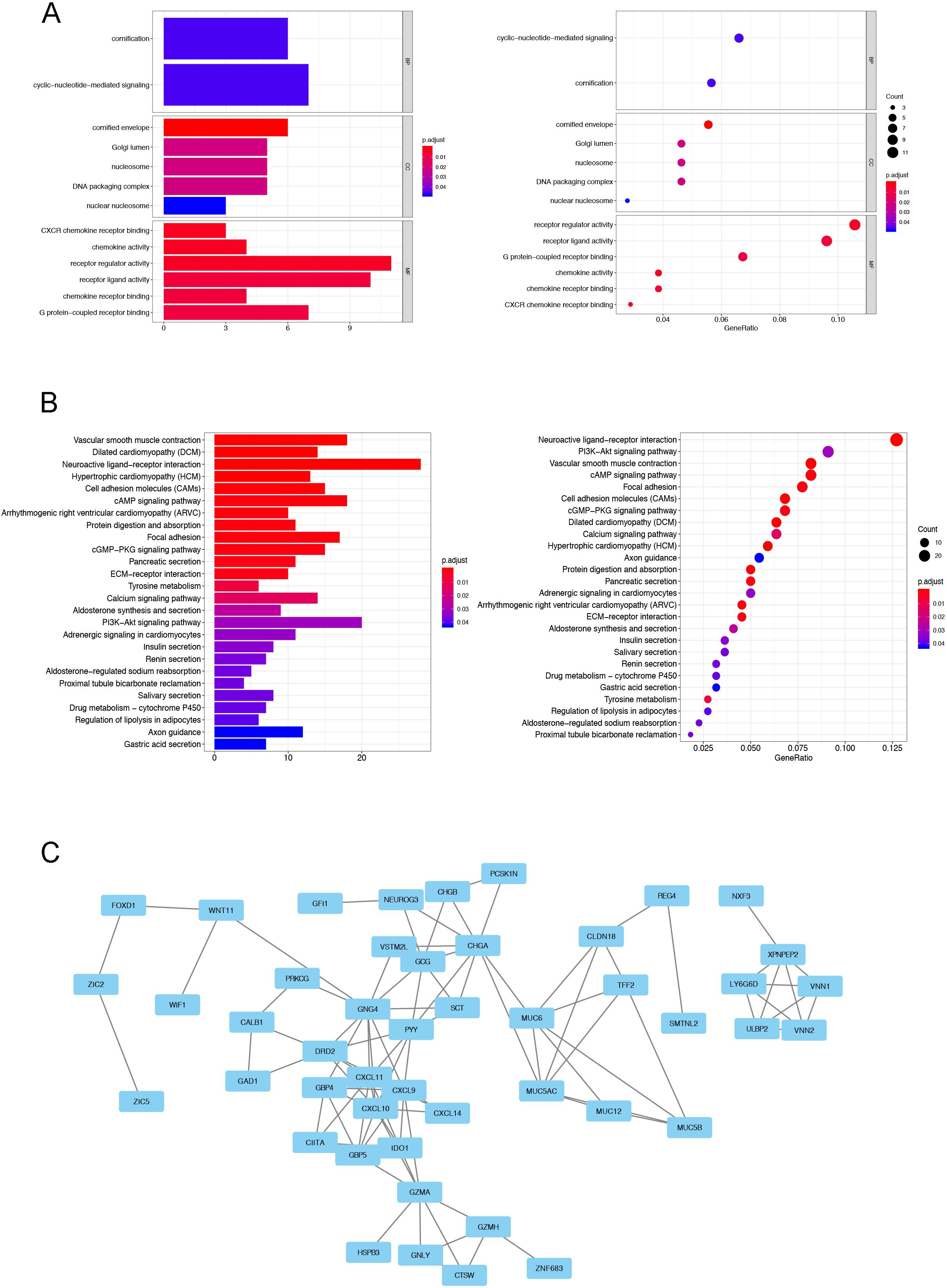
Then the Gene Onotology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed to evaluate the potential functions of these DEGs. The major components of enriched GO terms including CC (cellular component), BP (biological process), and MF (molecular function) ontologies. Our results indicated that the DEGs were mainly in the MF, such as CXCR chemokine receptor binding, chemokine activity, receptor regulator ligand activity, chemokine receptor binding, G protein-coupled receptor binding (Fig. 3A). In addition, the results of our KEGG enrichment analysis presented that the DEGs were involved in vascular smooth muscle contraction, dilated cardiomyopathy (DCM), neuroactive ligand-receptor interaction, hypertrophic cardiomyopathy (HCM), cell adhesion molecules (CAMs), cAMP signaling pathway, and protein digestion and absorption (Fig. 3B).
Protein and protein interaction (PPI) network is a kind of diagram to present the connectivity of the genes. Then we established a PPI network to identify the hub genes in the TMB related DEGs of CRC. Firstly, the connectivity degrees of the DEGs were calculated through a web-tool named STRING (string-db.org). Due to the evaluated degrees, the PPI network was constructed and the results were showed in Fig. 3C. Furthermore, we identified the 10 hub genes in this network, including G protein subunit gamma 4 (GNG4, degree
Expression patterns and Kaplan-Meier analysis of ten hub genes in CRC
Using the GEPIA web-tool, we accessed the expression patterns of 10 hub genes in the tumor tissues and normal tissues based on TCGA database. It was indicated that GNG4, CXCL11, CXCL10, CXCL9, and GZMA were up-regulated in CRC tumors as compared to normal tissues. Furthermore, comparing to normal tissues, CHGA, PPY, and GCG were down-regulated in CRC tissues. However, the expressions of GBP5 and DRD2 showed no significant differences between the CRC tissues and non-tumorous tissues (Fig. 4). And then we also carried out the Kaplan-Meier analysis to determine whether these hub genes could influence the prognosis of CRC patients. Our results illustrated that higher TGNG4 and GCG expression predicted the lower OS rates for the CRC patients (Fig. 5). In addition, following the increased expression of GNG4, CXCL11, CXCL10, and CXCL9, the TFS of CRC patients became low (Fig. S3). Then we also performed meta-analysis of GNG4 expression of CRC through Oncomine database. As shown in Fig. S4, in the 13 GEO datasets, about 8 studies presented that GNG4 was up-regulated in the CRC tissues. Combined these observations, GNG4 was confirmed to be up-regulated in tumor tissues and predicted the bad prognosis of CRC patients (Fig. S4A).
The 10 hub genes associated with TMB in CRC
The 10 hub genes associated with TMB in CRC
The expression patterns of 10 hub genes in CRC samples and non-cancerous samples based on TCGA database. In this part, the false discovery rate (FDR) was utilized to adjust the 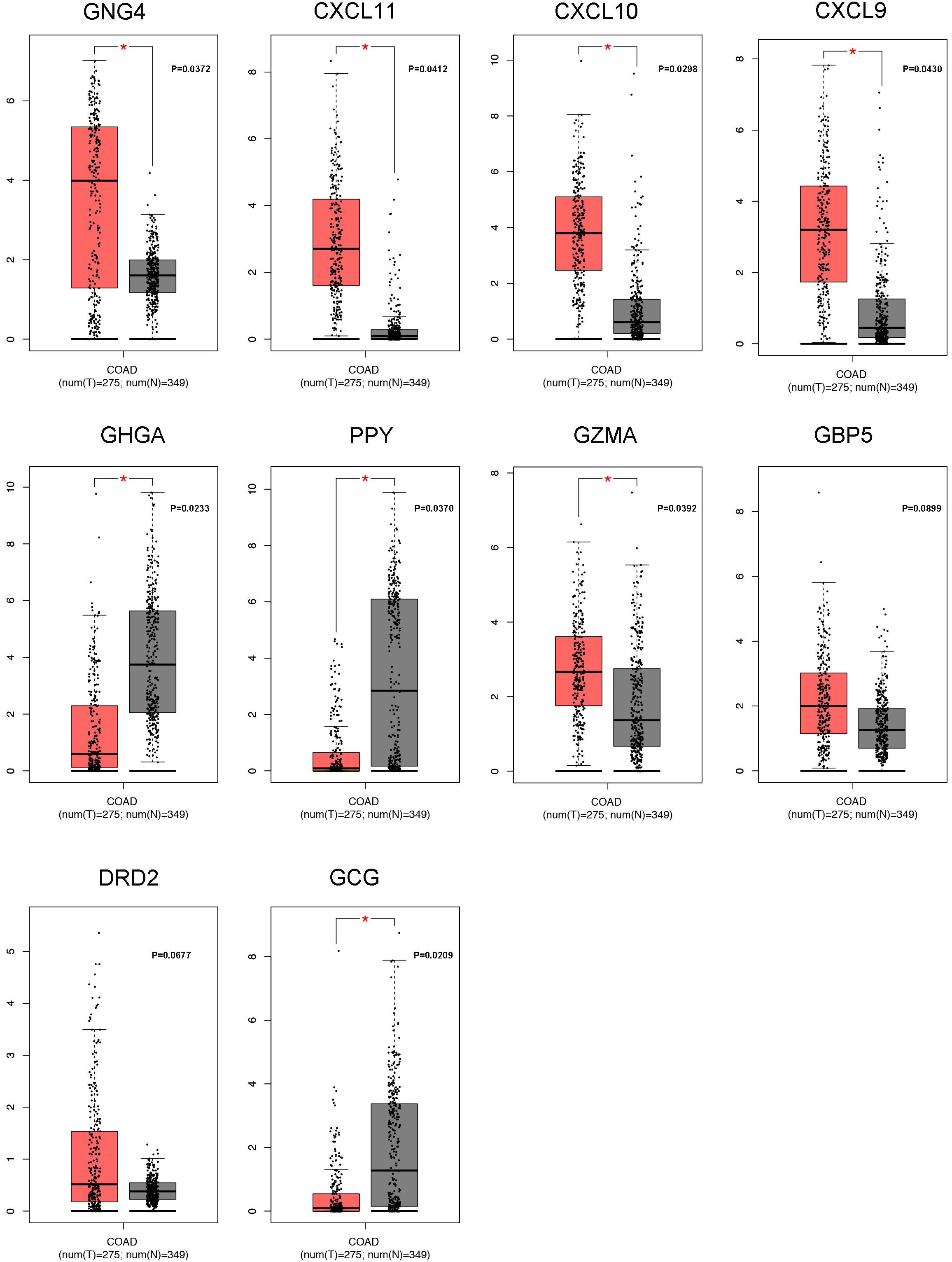
The Kaplan-Meier analysis of overall survival (OS) of ten hub genes in CRC patients. In this part, the false discovery rate (FDR) was utilized to adjust the 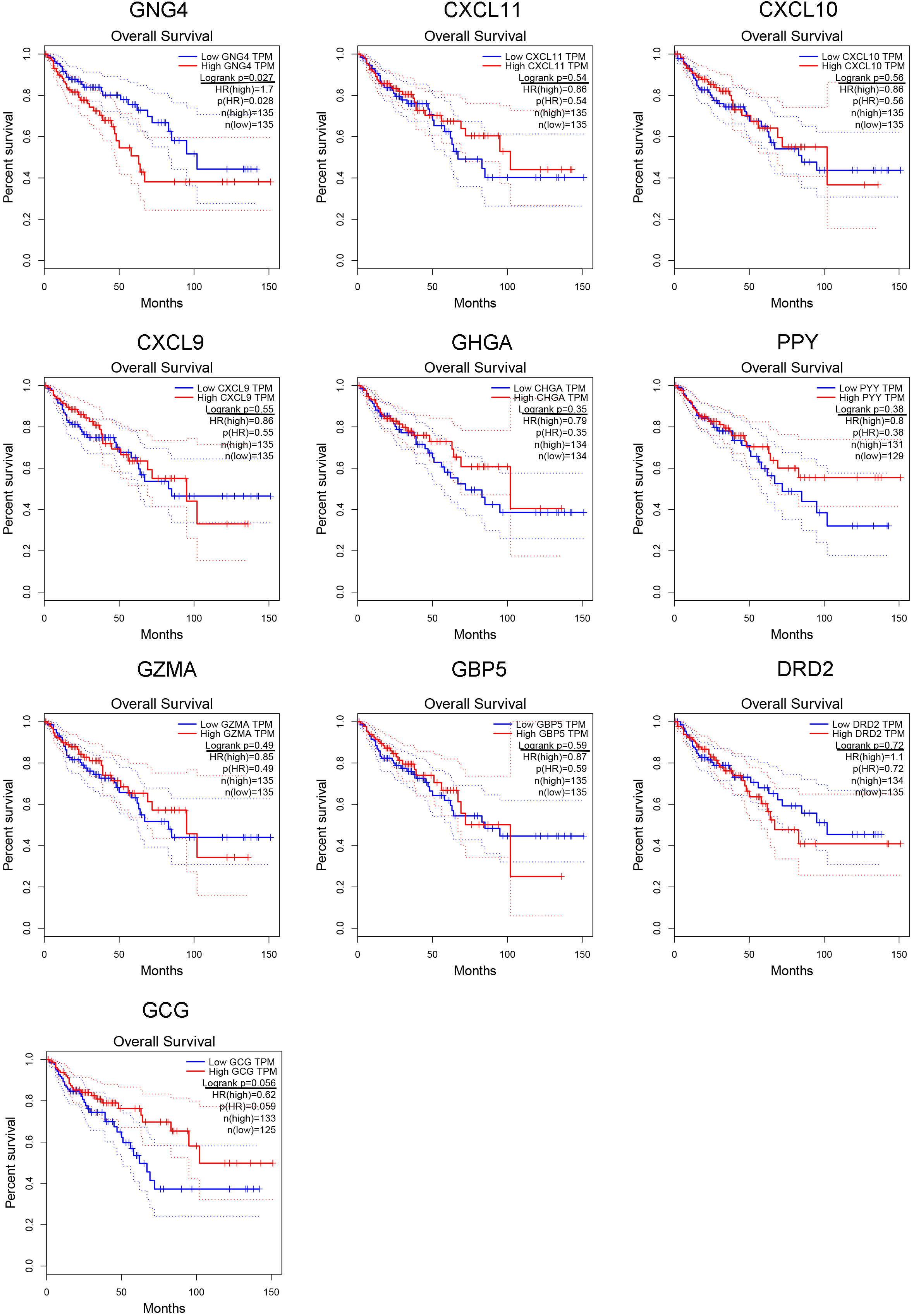
Most studies have revealed that CRC could invaded to the adjacent organs such as liver. Thus, we considered whether GNG4 could induce the metastasis of CRC into liver. The tissues of CRC and CRC tumor invaded to liver were then collected from our center. Using the immunohistochemical (IHC) staining, we evaluated the protein expression of GNG4 in these two kinds of tissues. As the Fig. S4B showed, GNG4 both presented higher expression in CRC and metastasis tumor as compared to normal colon tissue. Therefore, we thought that GNG4 might play an important role in colorectal cancer TMB and induce its metastasis in liver.
Using the CIBERSORT, we evaluated the distribution of 22 types of immune cells in TMB high group and TMB low group. And the 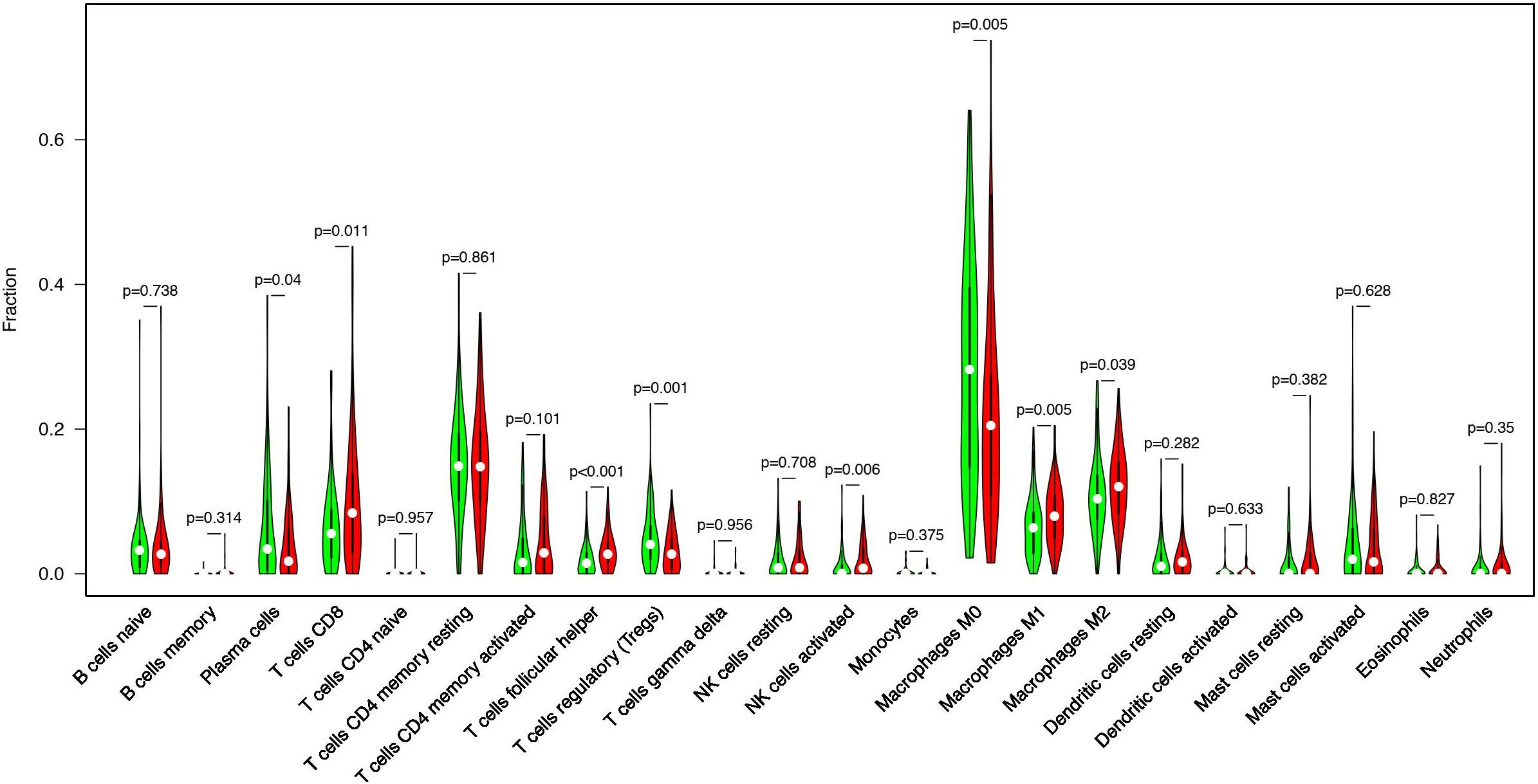
Increased tumor infiltrating lymphocytes (TILs) are highly of prognostic and predictive value for various kinds of tumors. CIBERSORT, a novel gene expression-based method, can estimate the levels of distinct leukocyte subtypes in tumors. Then we performed the CIBERSORT analysis to evaluate the abundance ratio of 22 immune cells in TCGA CRC samples. Our results demonstrated that the purity of plasma cells, T cells CD8, T cells follicular helper, T cells regulatory (Tregs), NK cells activated, macrophages M1, and macrophages M2 were differentially distributed between low TMB group and high TMB group of CRC patients (Fig. 6).
Discussion
In the recent studies, high tumor mutation burden (TMB) have been treated as an emerging selection biomarker for immune checkpoint blockade in tumors [14, 15]. As a novel immune related evaluation index, TMB is typically calculated from whole genome sequencing or whole exome sequencing (WES) data [16, 17]. In this study, we collected the WES and clinical data about CRC patients from TCGA database. Frequently mutated genes and hotspot mutations and their associations with TMB level were then systematically analyzed.
TMB is defined as the number of somatic coding mutations per million bases, and some genes were frequently mutated in CRC tumors. In many studies, the correlation between the TMB and CRC pathogenesis was observed. A recent study revealed that TMB-high was associated with better prognosis in patients with colorectal cancer treated with curative surgery followed by adjuvant fluoropyrimidine and oxaliplatin chemotherapy. This finding firstly confirmed that TMB would influence the effect of chemotherapy in CRC patients [18]. Besides, another study also indicated that a pan-cancer TMB panel could serve as a reference data-set for TMB-oriented panel design to identify CRC patients for immunotherapy [19]. In addition, Anqi Lin et al. found out that a mutation panel (including CREBBP, NOTCH3, PTCH1, CIC, DNMT1, SPEN) could affect the effects of immune checkpoint inhibitors in CRC [20]. The study carried out by Sachin G Pai et al. also observed that TMB-L may be a predictive biomarker in a subset of CRC patients treated with chemotherapy in CRC [21].
In our study we identified that TTN, APC, MUC16, SYNE1, TP53, KRAS53, FAT4, RYR2, PIK3CA, and ZFHX4 were the TOP 10 mutated genes in CRC. In addition, our findings illustrated that the overall survival of CRC patients became better followed by the higher level of TMB, and the TMB in stage I and stage II was higher as compared to the TMB in stage III and stage IV of CRC (
Then we tried to access the hub genes associated with TMB in the CRC tissues. The CRC tissues were divided into the low TMB group and high TMB group, and the DEGs were explored. And the subsequent KEGG and GO analysis were executed, we observed that these DEGs were involved in the immune associated pathways, such as CXCR chemokine receptor binding, chemokine activity, and receptor regulator ligand activity. Moreover, through the PPI analysis, 10 hub genes in this network were illustrated (GNG4, CXCL11, CXCL10, CXCL9, CHGA, PYY, GZMA, GBP5, DRD2, and GCG). Among this panel, we firstly confirmed that GNG4 was up-regulated in CRC tumors, and GNG4 was associated with the OS and TFS of CRC patients.
GNG4 is one of the fourteen
Conclusion
In this research, we identified that high TMB value was correlated to the good prognosis of CRC patients. Through the protein-protein interaction (PPI) analysis, we screened out the ten hub genes among the TMB related DEGs. Subsequently, we observed that GNG4 was up-regulated in tumor tissues, and GNG4 was related to the overall survival (OS) and tumor free survival (TFS) of CRC patients. Therefore, we considered GNG4 was correlated to the tumor immune microenvironment of CRC.
Footnotes
Acknowledgments
This study was supported by the Key project of Zhejiang Provincial Natural Science Foundation (No. LZ20H160002), Medical and Health Science and Technology Project of Zhejiang Province (2019KY290, 2021KY024), and National natural science foundation of China (No. 81800558).
Conflict of interest
The authors report no conflicts of interest in this work.
Supplementary data
Mutated genes involved in TMB of CRC tissues.
Heat-map presented the DEGs between the high TMB group and low TMB group in CRC. Red column symbolized for up-regulated genes. Green column symbolized for the down-regulated genes.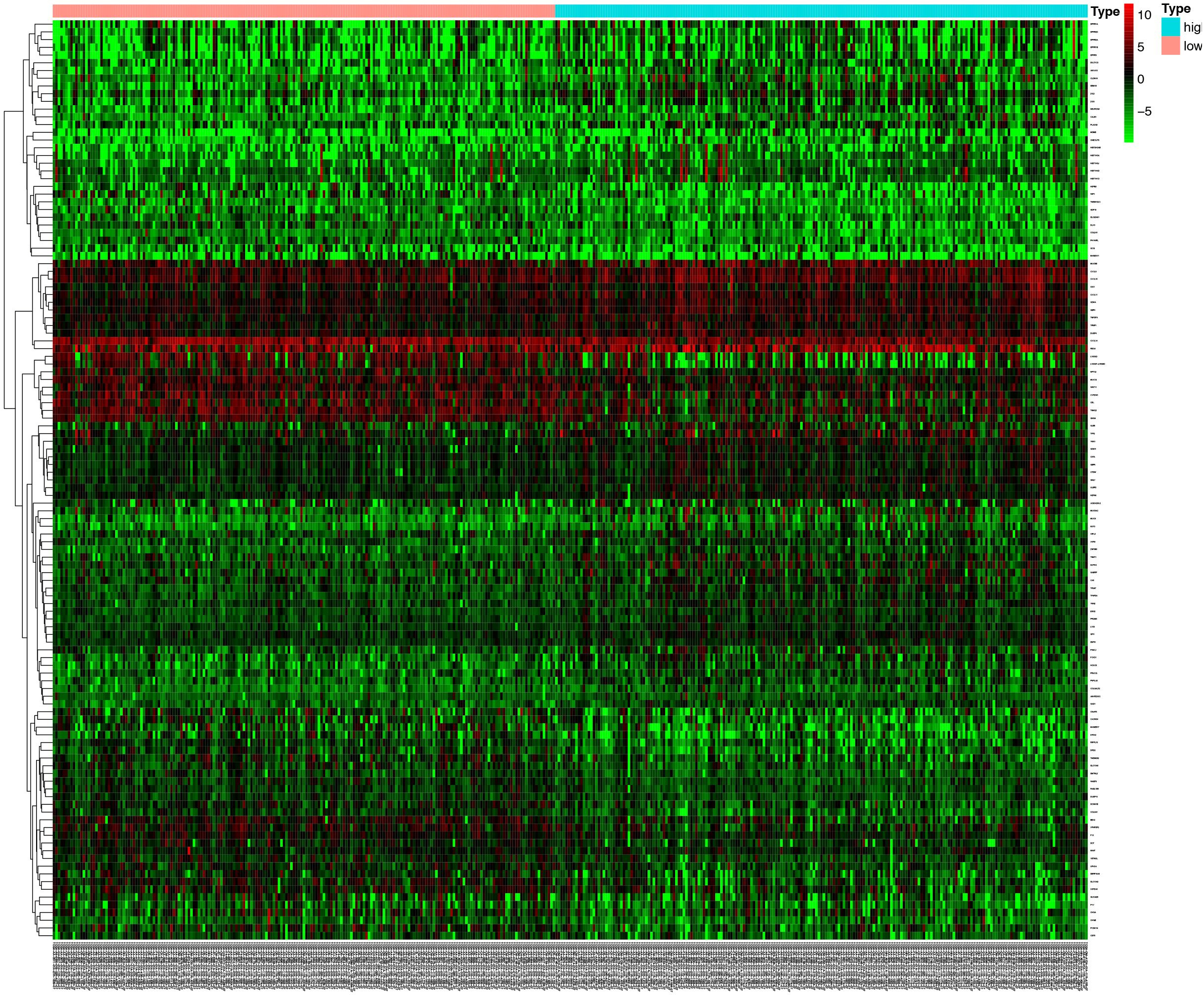
The Kaplan-Meier analysis of tumor free survival (TFS) of ten hub genes in CRC patients. In this part, the false discovery rate (FDR) was utilized to adjust the 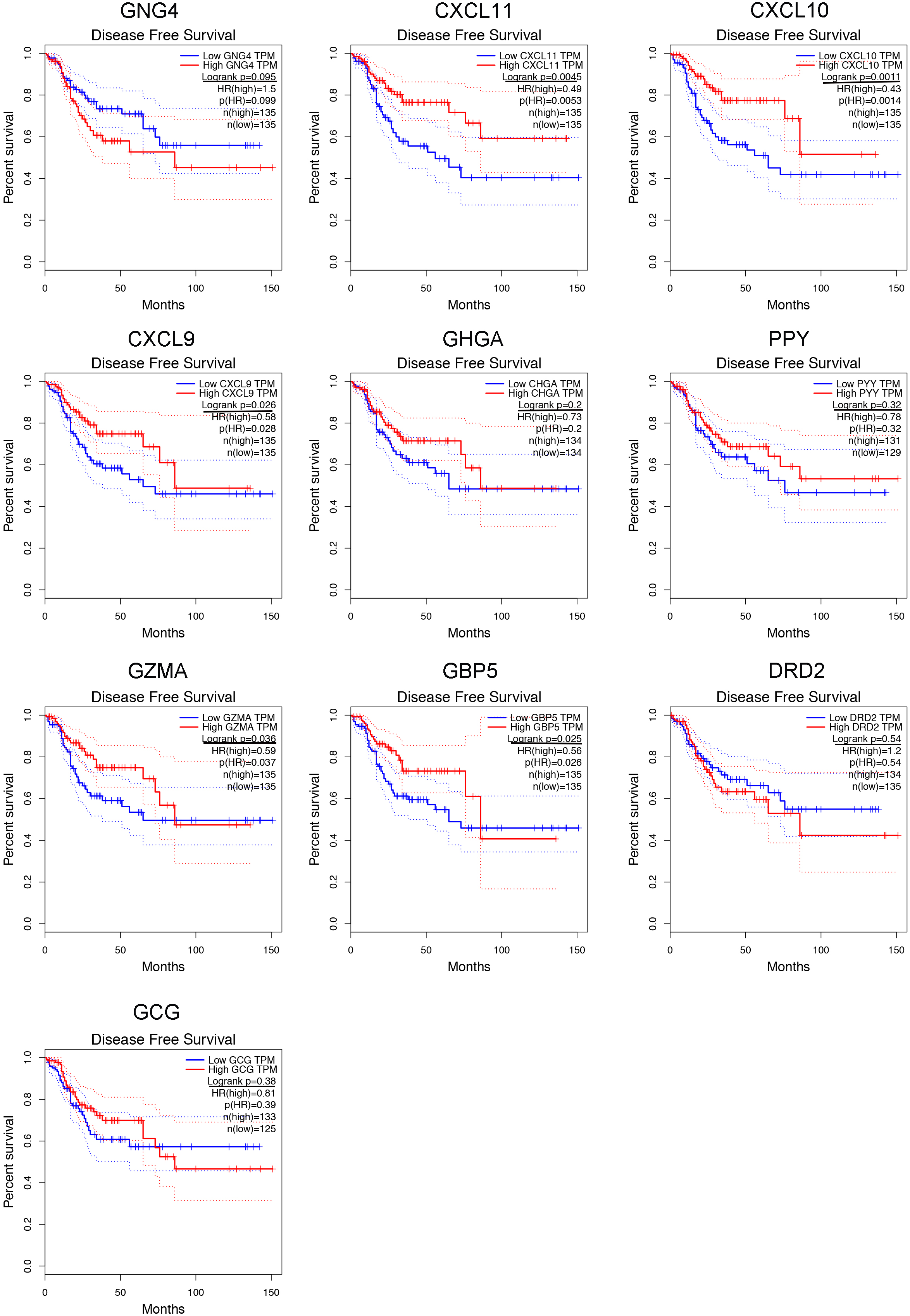
A. Meta-analysis of GNG4 expression of CRC through Oncomine database indicated shown that, in the 13 GEO datasets, about 8 studies presented that GNG4 was up-regulated in the CRC tissues. B. GNG4 both presented higher expression in CRC and metastasis tumor as compared to normal colon tissue.