
Abstract
BACKGROUND:
Breast cancer (BC) is the second most common cause of death from cancer in women in the United States. As the molecular mechanism of BC has not yet been completely discovered, identification of hub genes and pathways of this disease is of importance for revealing molecular mechanism of breast cancer initiation and progression.
OBJECTIVE:
This study aimed to identify potential biomarkers and survival analysis of hub genes for BC treatment.
METHODS:
The differentially expressed genes (DEGs) between breast cancer and normal cells were screened using microarray data obtained from the Gene Expression Omnibus (GEO) database. Gene ontology (GO) and KEGG pathway enrichment analyses were performed for DEGs using DAVID database, the protein-protein interaction (PPI) network was constructed using the Cytoscape software, and module analysis was performed using MCODE. Then, overall survival (OS) analysis of hub genes was performed by the Kaplan-Meier plotter online tool. Finally, the potential molecular agents were identified with Connectivity Map (cMap) database.
RESULTS:
A total of 585 DEGs were obtained, which were significantly enriched in the terms related to positive regulation of cell migration, regulation of cell proliferation and focal adhesion. KEGG pathway analysis showed that the significant pathways included Focal adhesion, Pathways in cancer, ECM-receptor interaction, Ribosome, Transcriptional misregulation in cancer and other signaling pathways about cancer. The PPI network was established with 576 nodes and 1943 edges. A significant module was found from the PPI network, the enriched functions and pathways included ECM-receptor interaction and Focal adhesion.
CONCLUSIONS:
Fifteen genes were selected as hub genes because of high degrees, among which, low expression of four genes was associated with worse OS of patients with BC, including RPS9, RPL11, RPS14 and RPL10A. Additionally, the small molecular agent emetine may be a potential drug for BC.
Keywords
Introduction
Breast cancer (BC) is the most common malignant tumor among females, and leads to the second most common cancer-related death in the United States. In 2017, there were 316,120 new cases and 40,610 deaths in the USA [1, 2]. As a multi-factorial disease, lack of exercise, late childbirth, obesity and other factors could cause BC [3]. The survival rate of breast cancer is closely related to the time of diagnosis. Overall, the average 5-year survival rate with breast cancer is 90%, but if the cancer has distant metastasis, the 5-year survival rate drops down to 26% [2].
According to clinical and histological manifestations of breast cancer, it can be classified into different subtypes [4]. If patients with breast cancer are diagnosed and treated at early time, they will have better survival probability. Generally, many factors may affect treatment plan including the stage of the disease at diagnosis; the status of the human epidermal growth factor receptor 2 (HER2) and the hormone receptor; health status; the occurrence of gene mutation, age, and whether the patient has experienced menopause [2]. As we all know, triple-negative breast cancer does not express estrogen receptors (ER), progesterone receptors (PR), and/or HER2, but expresses receptors for other hormones [5]. BC is a highly lethal tumor and ranks second in the most susceptible tumor among women, but the molecular mechanisms of BC occurrence and progression are not fully understood. Therefore, it would be worthwhile to clarify the potential molecular mechanisms of BC for the purpose of identifying new biomarkers and discover candidate small molecular drugs.
In the study, we employed the bioinformatics approach to identify DEGs between breast tumor and normal control samples. In addition, other methods were performed to analyze this data, including hierarchical clustering analysis, GO/KEGG pathway analysis, construction of protein-protein interaction network, sub-module analysis, survival analysis and identification of small molecular agents, to identify key genes and pathways in BC. The aim of this study was to gain a more in-depth understanding of the mechanisms and to find biomarkers and potential therapeutic targets for BC.
Materials and methods
Data collection
The gene expression profile of 28 breast cancer samples and 5 normal samples (GSE10797) based on the platform of GPL571 was collected from GEO database (
Identification of DEGs
RMA [7] algorithm in Affy [8] package was applied to preprocess the raw expression data in the R statistical software. Then, we analyzed DEGs between BC and normal samples using significance analysis of the microarrays method with limma package [9], following the criteria
GO term and KEGG pathway analysis
GO is used to perform functional studies on gene sets [11]. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database is used to understand the advanced features and biological systems (such as cell, biological and ecological systems) [12]. The Database for Annotation, Visualization and Integrated Discovery (DAVID) is a biological database, which integrates biological data and analysis tools for a large list of genes [13, 14].
Heat map of the top 100 DEGs. Red: high expression level; green: low expression level.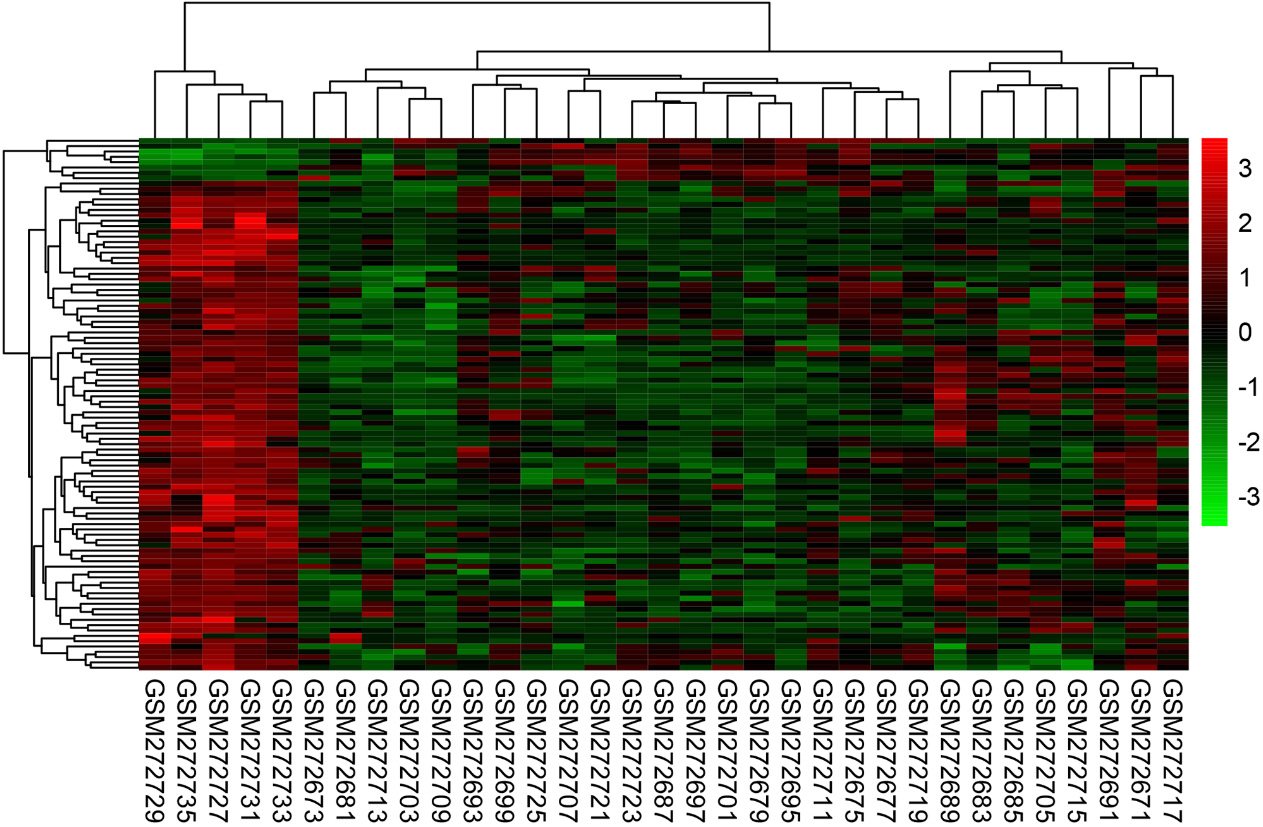
GO enrichment analysis result of DEGs. BP biological process, CC cellular component, MF molecular function.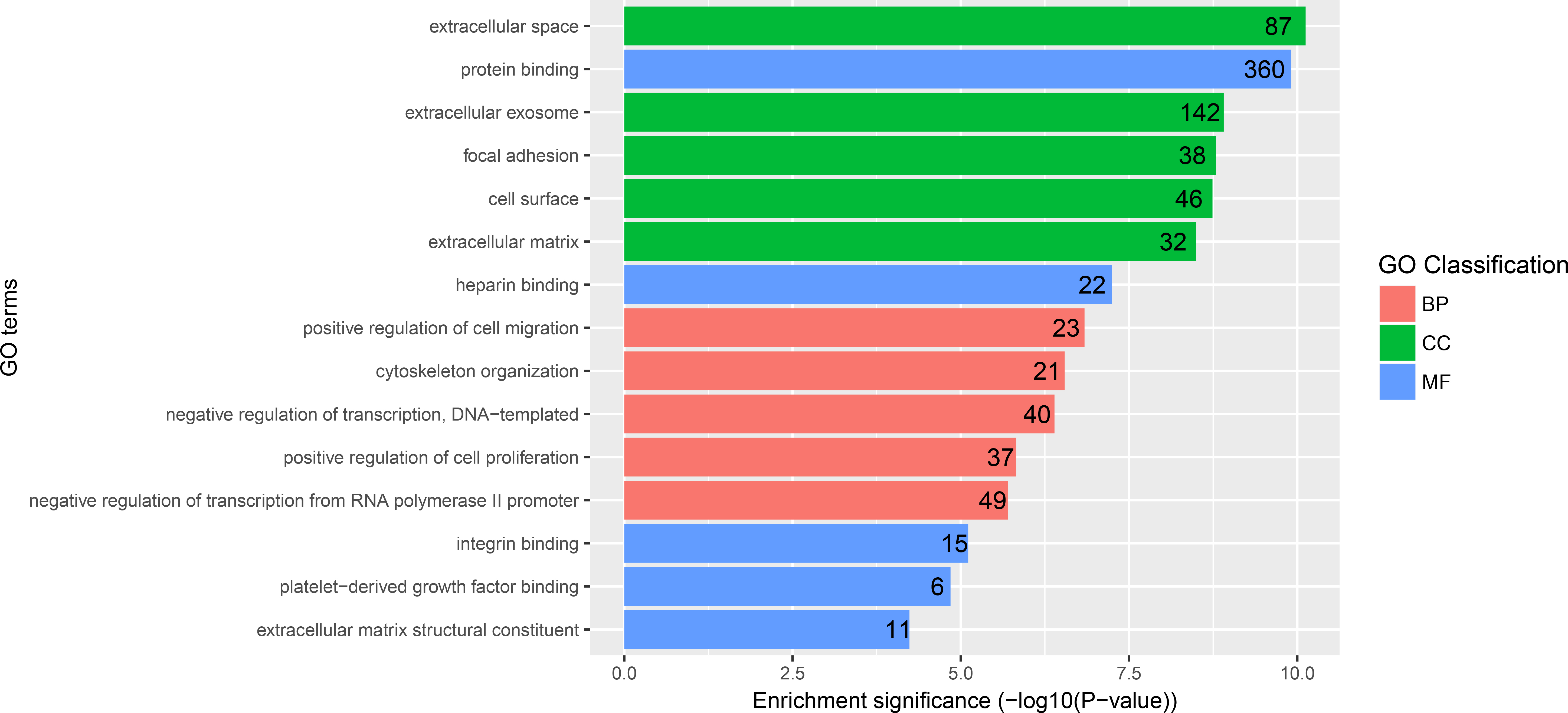
Search Tool for the Retrieval of Interacting Genes (STRING), a database of protein prediction function correlation [15]. We used Cytoscape to visualize significant gene pairs in the PPI network [16]. The combined score of
Survival analysis of hub genes
Kaplan-Meier plotter (
Identification of small molecular drugs
The Connectivity Map (cMap) [19] is a public database, which collects of more than 7,000 expression profiles from cultured human cells treated with small molecules. The DEGs in PPI network were mapped onto the cMap database. The
GO and KEGG analysis of the genes in module
GO and KEGG analysis of the genes in module
KEGG pathways analysis result of DEGs.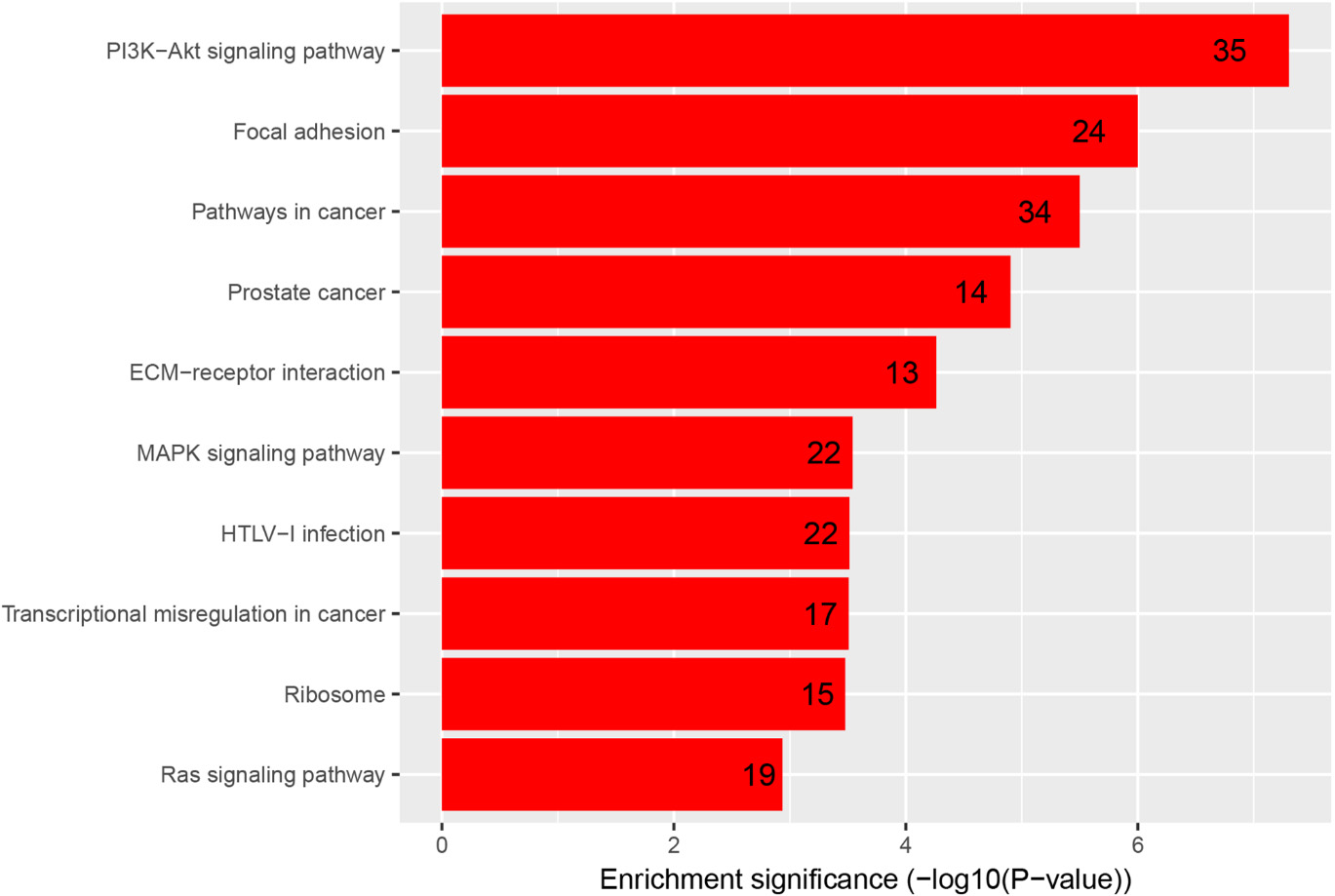
PPI network and a significant module. A. PPI network of DEGs. B. A significant module selected from PPI network. Red nodes represent up-regulated genes; green nodes represent down-regulated genes.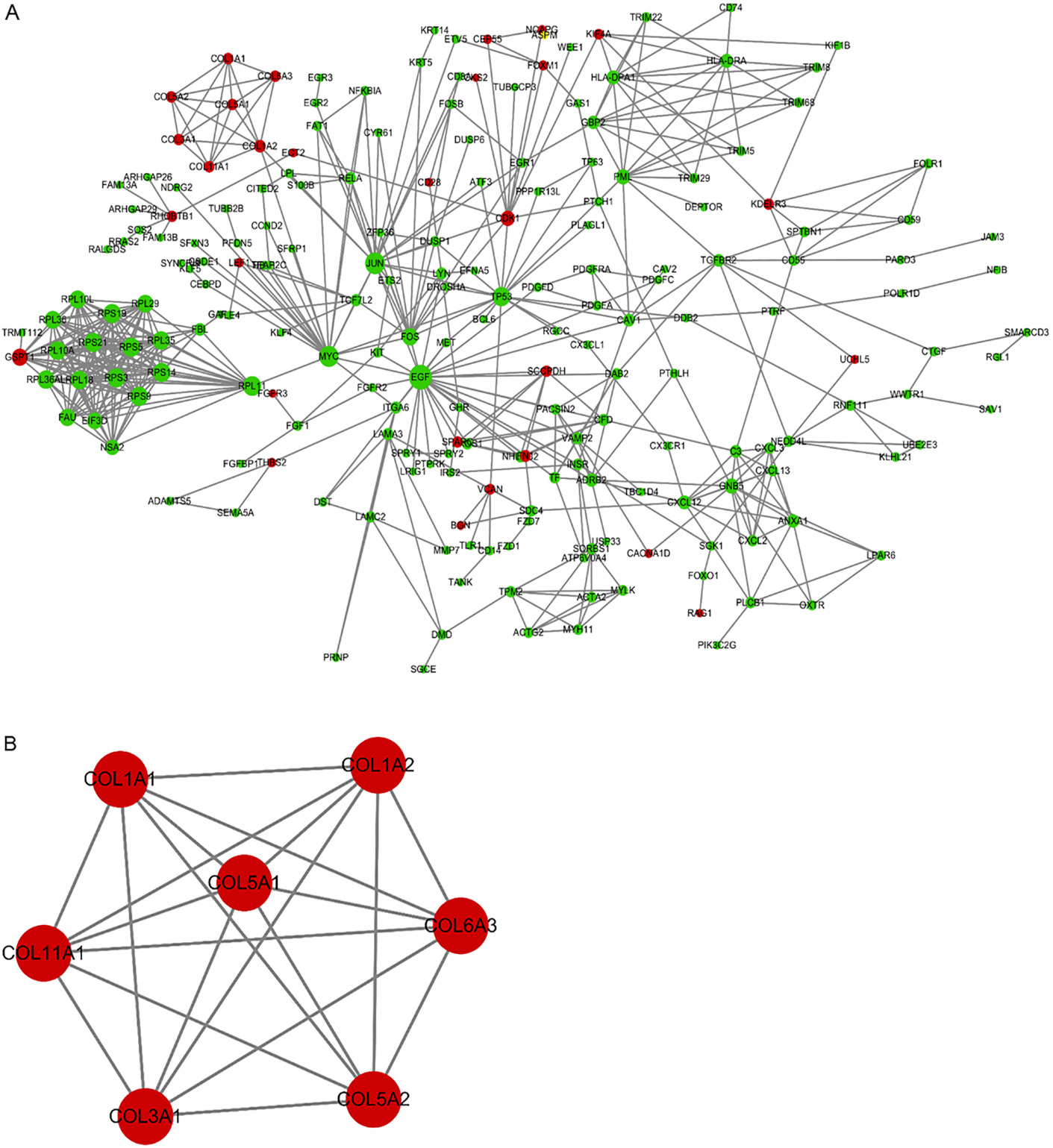
The identification of DEGs
Gene expression dataset GSE10797 was got from GEO database, including 28 breast cancer samples and 5 normal samples. After gene differential expression analysis of microarray data, 585 genes were identified to be differentially expressed genes, among which 71 genes were upregulated and 514 genes were downregulated in tumor tissues. The expression level of the top 100 DEGs was displayed in Fig. 1.
GO term enrichment analysis of DEGs
According to the distribution of DEGs in the Gene Ontology, the gene function of DEGs were clarified. The top five GO terms of DEGs were shown in Fig. 2. As to biological process, the DEGs were significantly enriched in positive regulation of cell migration, regulation of cell proliferation, cytoskeleton organization, negative regulation of transcription, DNA-templated, and negative regulation of transcription from RNA polymerase II promoter. For cellular component, the DEGs were significantly enriched in focal adhesion, extracellular exosome, extracellular space, cell surface, and extracellular matrix. About molecular function, the DEGs were significantly enriched in protein binding, heparin binding, integrin binding, platelet-derived growth factor binding and extracellular matrix structural constituent.
KEGG pathway analysis of DEGs
As shown in Fig. 3, KEGG pathways results showed that the DEGs were significantly enriched in Focal adhesion, Pathways in cancer, ECM-receptor interaction, Ribosome, Transcriptional misregulation in cancer and other signaling pathways about cancer development. These significantly enriched terms and pathways will provide further insight toward future research directions about the role which DEGs play in BC occurrence and progress.
PPI network analysis of DEGs and modules selection
The PPI network of DEGs was composed of
Kaplan-Meier plot of overall survival using four genes in breast cancer patients. Prognostic value of RPS9 (a), RPL11 (b), RPS14 (c) and RPL10A (d) were obtained in 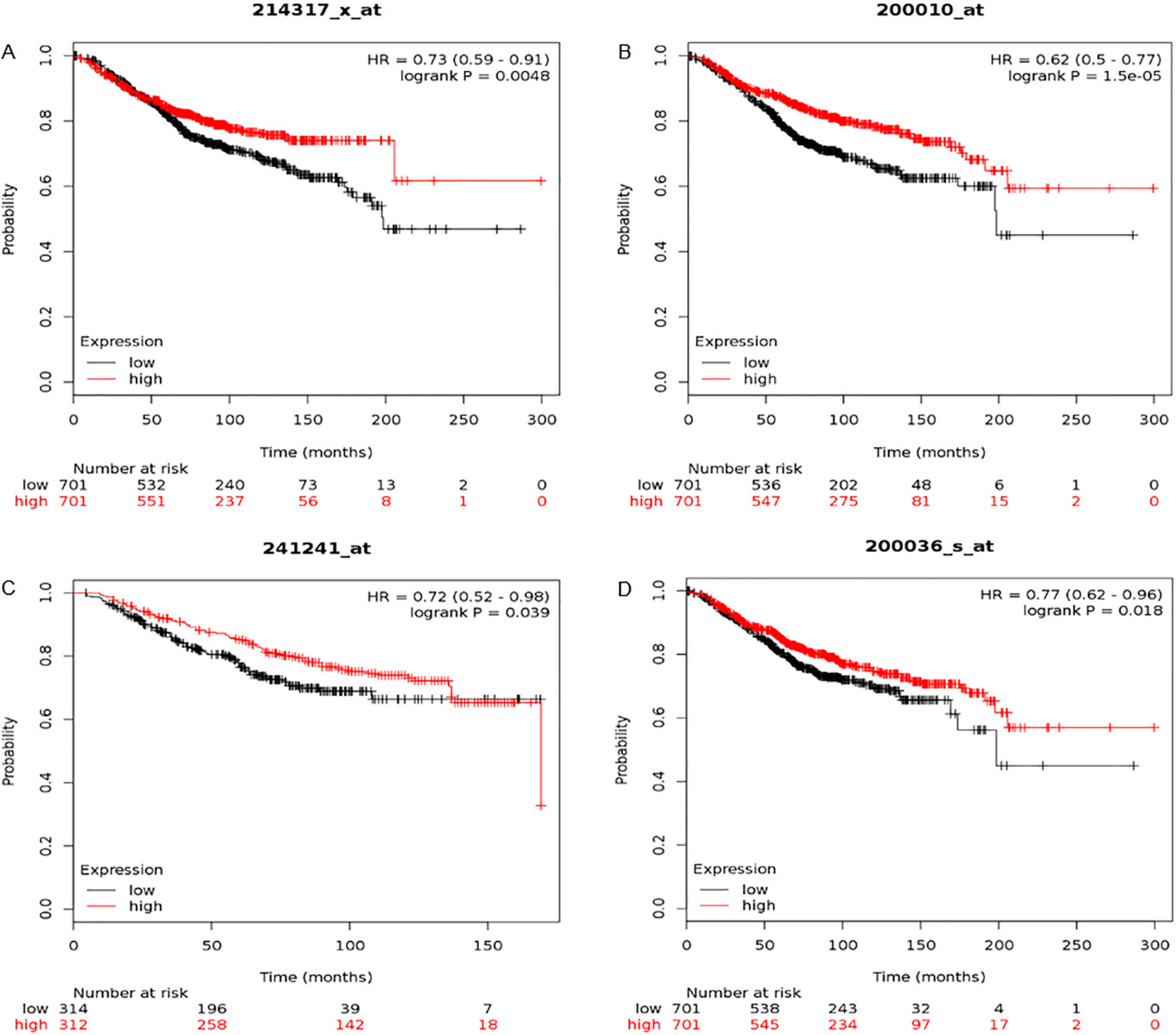
Enriched significant of small molecules
As for 15 hub genes, the prognostic value of them was analyzed in Kaplan-Meier plotter. Overall survival for BC patient was got by the expression of each gene. It showed that low mRNA expression of RPL11 was related with worse overall survival, as well as RPS14, RPS9 and RPL10A (Fig. 5).
Identification of small molecular agents
Based on the results of cMap database mapping, a total of 10 small molecular agents were obtained, such as emetine and cephaeline. In Table 2, the small molecular agent emetine had the highest negative score.
Discussion
In this study, we got 585 DEGs, including 71 up-regulated genes and 514 down-regulated genes in BC samples. These DEGs were mainly enriched in several functional terms such as positive regulation of cell migration, regulation of cell proliferation, focal adhesion, protein binding, and KEGG pathways including Focal adhesion, Pathways in cancer, ECM-receptor interaction, Ribosome, Transcriptional misregulation in cancer and other signaling pathways about cancer development. Among these DEGs, we found 15 hub genes in the PPI network. Then, RPS9, RPL11, RPS14 and RPL10A, four down-regulated genes were significantly correlated with worse overall survival of breast cancer samples by survival analysis. In addition, small molecular agent emetine was identified as an important potential drug for breast cancer.
RPS9, RPL11, RPS14 and RPL10A are all Ribosomal Protein gene, among which, RPS9 and RPS14 encode a ribosomal protein that is a component of the 40S subunit, RPL11 and RPL10A encode a ribosomal protein that is a component of the 60S subunit. As we all know, ribosome plays a vital role in protein synthesis by protein translation and is also essential for cell growth, proliferation, and development. Recent study showed that somatic mutations and deletions of ribosomal protein genes were found in leukemias and solid tumors [20].
To date, the detailed function and role of RPS9 for breast cancer has yet been reported. However, decreased expression of RPS9 was found in many different cancers, including breast cancer, pancreatic cancer, and malignant astrocytic gliomas [21, 22, 23]. RPL11 inhibits the ability of HDM2 to degrade p53 and expression of RPL11 induces a p53 response [24]. When RPL11 was down regulated, HDM2 may inactive p53, thereby leading to tumor development. Meanwhile, low expression of RPL11 enhances c-Myc transactivation activity and cell proliferation [25]. And, overexpression of c-Myc is found in various human tumors [26, 27]. Similarly, reduction of endogenous RPL11 by siRNA increases c-Myc activities [25], which leads to cell growth, proliferation, tumorigenesis [28, 29, 30]. RPS14 may play a role in inhibiting tumor, because it can activate p53 and also inhibit c-Myc [31]. Because RPS14 and RPL11 have different binding patterns on MDM2 or c-Myc, so they may work together to suppress MDM2 and c-Myc activity [24, 25, 32]. Previous studies also have reported that the inactivation of RPS14 is apt to cell transformation [33]. Furthermore, RPL10A may be related to cell proliferation in view of its roles in embryogenesis and organogenesis [34, 35], and identification as a tumor antigen [36]. Lee et al. found RPL10 A gene was downregulated in glioblastoma [37]. In summary, this data suggests that RPS9, RPL11, RPS14 and RPL10A involve in the cell growth, proliferation and tumorigenesis, play a crucial role in the cancer development, which support our findings.
Furthermore, emetine was identified to be important small molecular agent in the development of BC. Emetine, a powerful inhibitor of protein synthesis in eukaryotes, has been shown to have potential for anticancer effects for cancer treatment [38, 39]. Emetine inhibits protein synthesis, which can effectively cause cancer cells to exogenous cell death pathways [40]. There was some evidence showed that Emetine was capable of inducing apoptosis and inhibiting proliferation in many different tumor cells [41, 42, 43, 44, 45], including bladder cancer and prostate cancer cell [43]. Emetine has cytotoxic effect on cancer cells because it can inhibit protein synthesis in eukaryotic cell ribosomes and interact with DNA [46]. Interestingly, a previous bioinformatics network-based approach has also identified emetine as candidate drug for treating BC [47]. These observations, together with the reported anti-cancer properties of emetine [48], strongly support emetine as a novel drug for the treatment of BC.
As a summary, the aim of the current study was to identify DEGs with integrated bioinformatics analysis, discover the potential biomarkers and predict progression of diseases. In our study, a total of 585 DEGs were screened out, and RPS9, RPL11, RPS14 and RPL10A might be potential biomarkers in breast cancer. Emetine may be a candidate drug for treating BC. Our results suggested that data mining and integration could be a useful tool to predict progression of breast cancer, to understand the mechanism of the occurrence and development of tumor. However, the present study was performed using bioinformatics methods and the conclusions remain to be confirmed by corresponding experiments. Therefore, further experimental study is required to verify the findings of the present result.
Footnotes
Acknowledgments
We thank Daoming Wang (University of Chinese Academy of Sciences) for his contribution in Fig. 2 presentation and Kang Shao (BGI-Shenzhen) for his contribution in language modify. This study was supported by the National Key Research and Development Program of China (2016YFC0902301).
Conflict of interest
The authors have no conflict of interest to declare.