
Abstract
BACKGROUND:
Prostate cancer (PCa) is the most common and the second leading cause of cancer-related death among men in America. As the molecular mechanism of PCa has not yet been completely discovered, identification of hub genes and potential drug of this disease is an important area of research that could provide new insights into exploring the mechanisms underlying PCa.
OBJECTIVE:
The aim of this study was to identify potential biomarkers and novel drug for prostate cancer treatment.
METHODS:
The differentially expressed genes (DEGs) between prostate cancer and normal cells were screened using microarray data obtained from the Gene Expression Omnibus database. Gene ontology (GO) and pathway enrichment analyses were performed in order to investigate the functions of DEGs, and the protein-protein interaction (PPI) network of the DEGs was constructed using the Cytoscape software. DEGs were then mapped to the connectivity map database to identify molecular agents associated with the underlying mechanisms of PCa.
RESULTS:
Totally, 359 genes (155 upregulated and 204 downregulated genes) were found to be differentially expressed between prostate cancer and normal cells. The GO terms significantly enriched by DEGs included cell adhesion, protein binding involved in cell-cell adhesion, response to BMP, extracellular region and extracellular region part. KEGG pathway analysis showed that the most significant pathways included cell adhesion molecules (CAMs) and TGF-beta signaling pathway. The PPI network of up-regulated DEGs and down-regulated DEGs were established, respectively. While CDH1, BMP2, NKX3-1, PPARG and PRKAR2B were identified as the hub genes in the PPI network.
CONCLUSIONS:
The BMP2, PPARG and PRKAR2B genes may therefore be potential biomarkers in the treatment of PCa. Additionally, the small molecular agent phenoxybenzamine may be a potential drug for PCa.
Introduction
According to WHO, prostate cancer ranks second in the most incident cancer, and ranks fifth in the key cause of death by cancer among men in the world [1]. As a multi-factorial disease, many factors including age, ethnicity, diet, and geographic factors are supposed to play a key role in PCa development [2]. In North America, PCa remains the most common noncutaneous solid tumor. It ranks third and second in death caused by cancer among men in Canada and USA [3, 4]. In 2016, a total of 180,890 new cases and 26,120 deaths from the cancer are expected to occur in the United States [5]. Overall, the five-year survival rate is excellent (98.9%) but drops down to 30% in the case of aggressive form with distant metastasis and relapse after treatment [1].
As for the early detection of PCa, prostate-specific antigen (PSA) is the most widely used biomarker, but it has poor specificity (33%) resulting in a high number of positive reports. So, PSA does not represent an ideal biomarker. To date, and despite the high number of studies, only very few biomarkers for the prediction of PCa progression have been verified in different cohorts or using different techniques, yet none was translated into clinical practice [6]. Moreover, the molecular mechanism of PCa occurrence and progression remains unclarified. Therefore, it would be worthwhile to clarify the potential molecular mechanisms of PCa for the purpose of identifying new biomarkers and discovering candidate small molecular drugs.
In our study, the differentially expressed genes between prostate tumor and matched normal tissues were identified by the bioinformatics approach. Additionally, several methods including hierarchical clustering analysis, GO/KEGG pathway analysis, construction of protein-protein interaction network and identification of small molecules were used to analyze this data. The aim of this study was to produce a systematic view on understanding the underlying mechanisms and to investigate potential therapeutic targets for prostate cancer.
Cartridge of expression values data before and after normalization. The x coordinate: the samples; the y coordinate: the gene expression values. Left: unnormalized data; right: normalized data.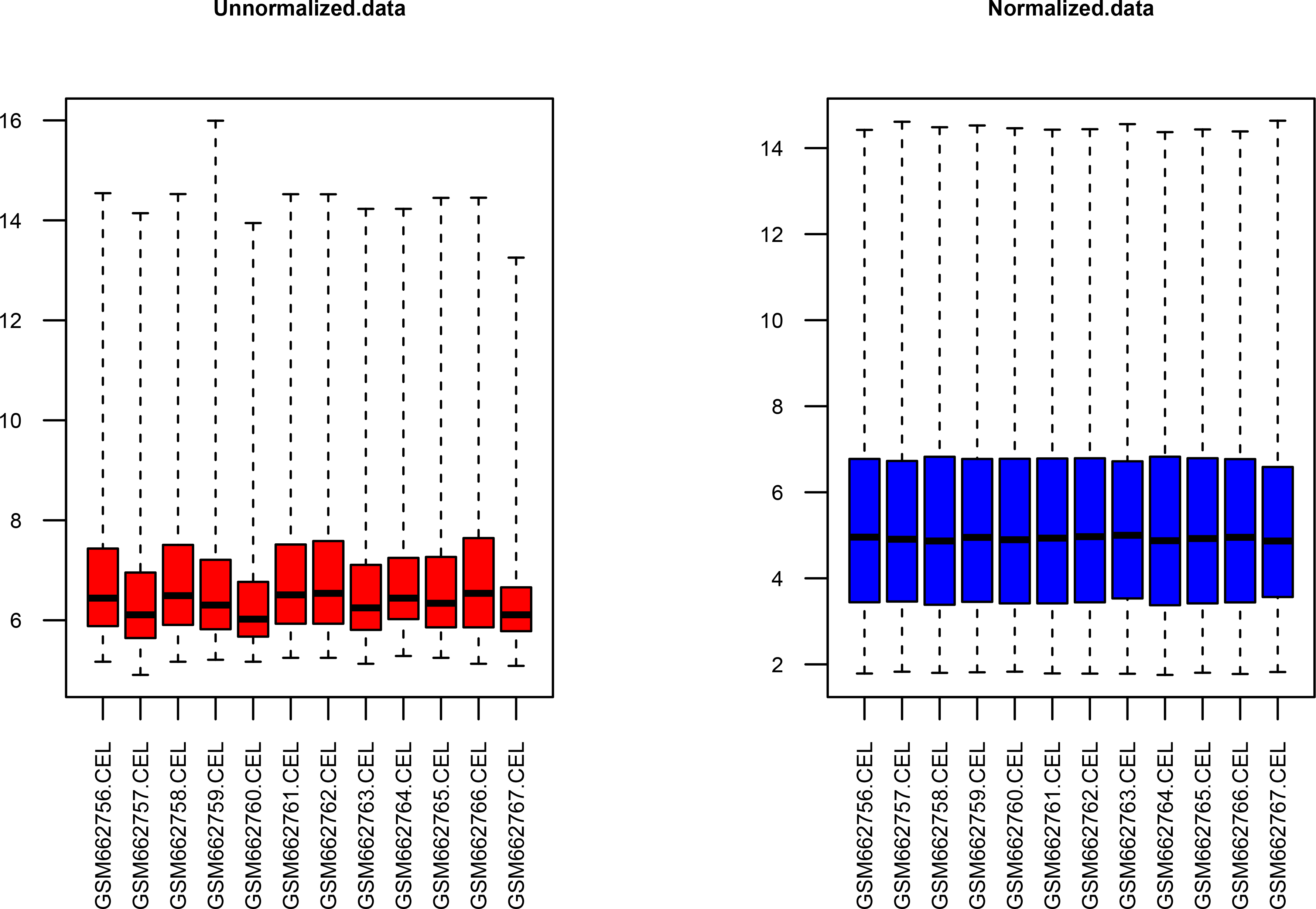
Heat map and clustering analysis of DEGs. The left of the heat map shows clustering of the DEGs, the above of the heat map shows clustering of the samples. Red: high expression level; green: low expression level.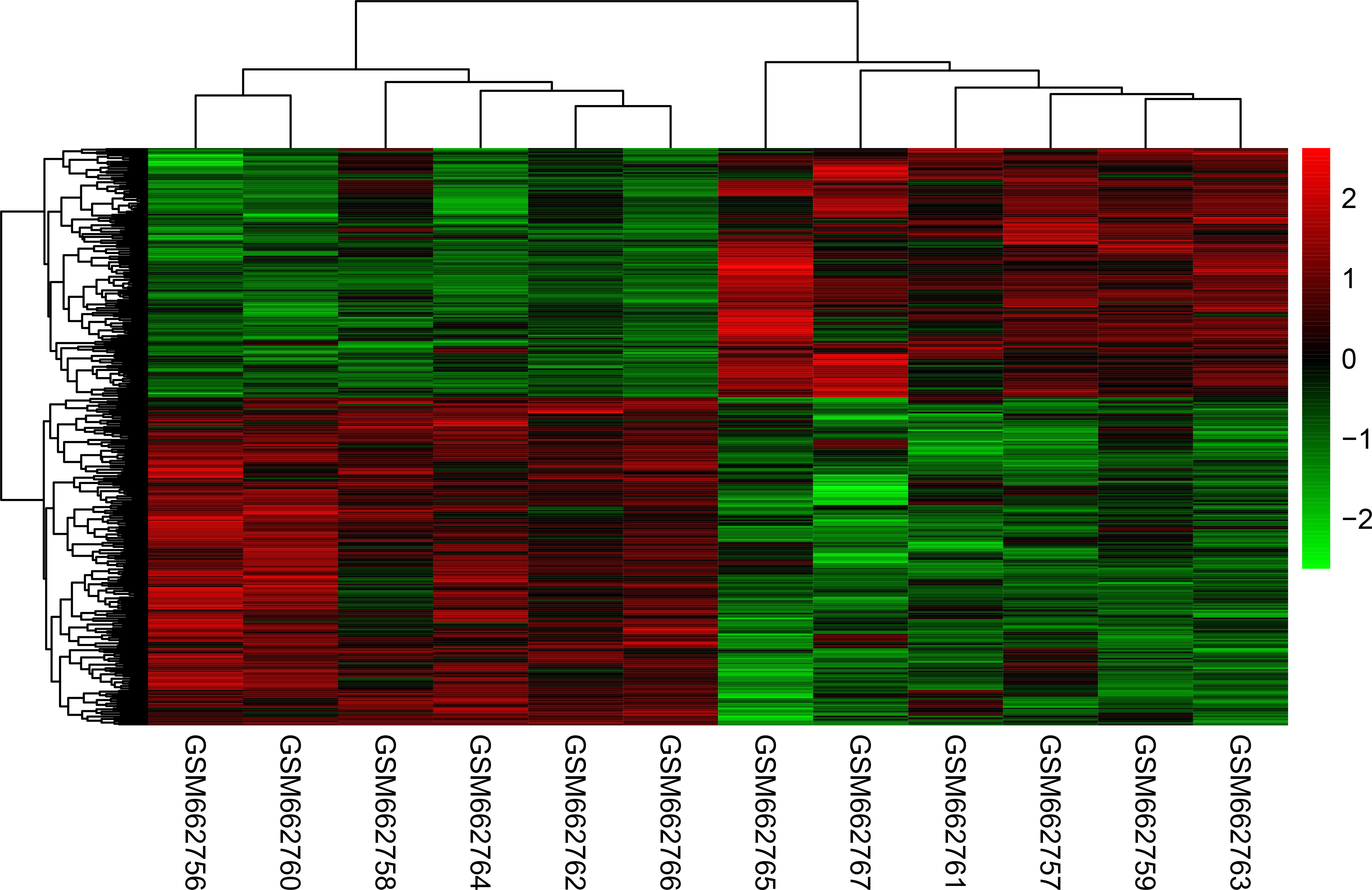
Data collection
The gene expression profile of six primary prostate tumors samples and six normal prostate tissues samples (GSE26910) based on the platform of GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array) was collected from the Gene Expression Omnibus (GEO;
The top 5 GO terms and KEGG pathway analysis for DEGs
The top 5 GO terms and KEGG pathway analysis for DEGs
The Affy package [8] was applied to preprocess the raw expression data with the rma [9] algorithm in the R statistical software. Then, the limma package [10] was used to identify DEGs between PCa and matched normal samples. We selected differentially expressed genes with
GO and KEGG pathway analysis
GO is used to perform functional enrichment analysis on gene sets [12]. Additionally, the Kyoto Encyclopedia of Genes and Genomes database is used to understand the high-level functions and utilities of the biological system [13]. The Database for Annotation, Visualization and Integration Discovery (DAVID) online tool [14, 15] was used for DEGs analysis of GO annotation and KEGG pathway enrichment.
Construction of PPI network
Search Tool for the Retrieval of Interacting Genes (STRING), a biological database and web resource, offers comprehensive information of predicted protein-protein interactions [16]. To evaluate the interactive relationship of DEGs, we mapped the DEGs to STRING. The combined score of
Identification of small molecular drugs
The Connectivity Map (cMap) [18] database is a collection of more than 7,000 expression profiles from cultured human cells treated with small molecules. The DEGs in PPI network were mapped onto the cMap database. The
Results
Data processing and DEGs analysis
The results of data processed before and after normalization were shown in Fig. 1, and the lines in the boxes were nearly on the same straight line after standardization, showing the standardization level was satisfactory. Then, 359 genes were identified to be differentially expressed genes, including 155 upregulated genes and 204 downregulated genes in tumor tissue.
As shown in Fig. 2, DEGs were divided into 2 clusters according to the clustering analysis result. Meanwhile, tumor and normal tissues were also classified into 2 different groups.
GO and KEGG pathway analysis
The top five GO terms of up-regulated and down-regulated DEGs were shown in Table 1. The upregulated DEGs were mainly enriched in cell adhesion, extracellular region part, cell-cell junction, cell adhesion molecule binding and protein binding involved in cell-cell adhesion, and the downregulated DEGs mainly participated in cell development, response to BMP, extracellular region, extracellular region part, extracellular space, receptor binding and sulfur compound binding.
In Table 1, the pathways results showed that the up-regulated DEGs were significantly enriched in cell adhesion molecules (CAMs) pathway, while the downregulated DEGs were significantly enriched in TGF-beta signaling pathway, Drug metabolism-cytochrome P450, Axon guidance and Hypertrophic cardiomyopathy (HCM) (Table 1).
The Protein network was constructed by online software string.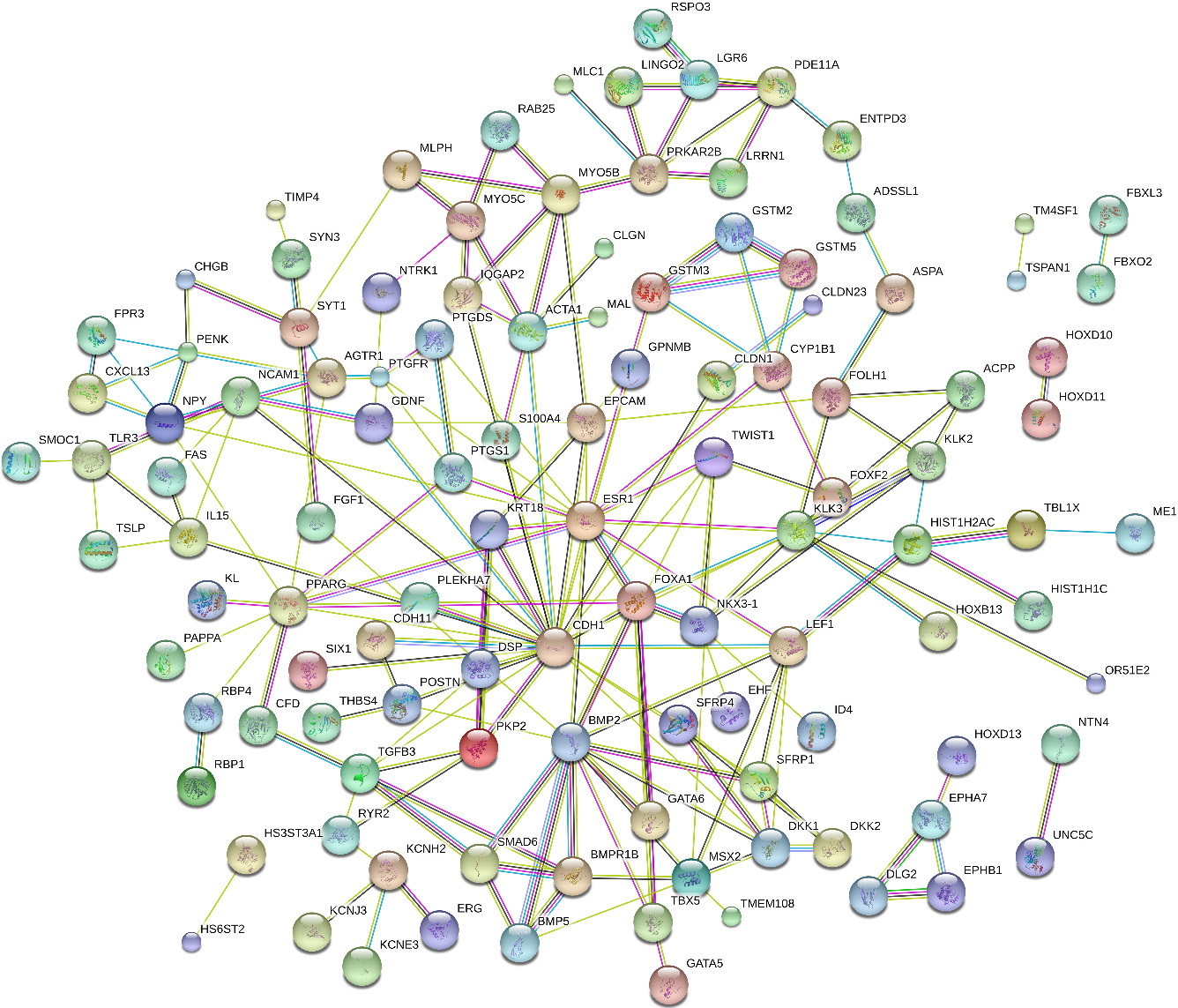
Constructed PPI network of DEGs. A: The network of up-regulated DEGs. Pink nodes represent upregulated genes; B: The network of down-regulated DEGs. Green nodes represent downregulated genes.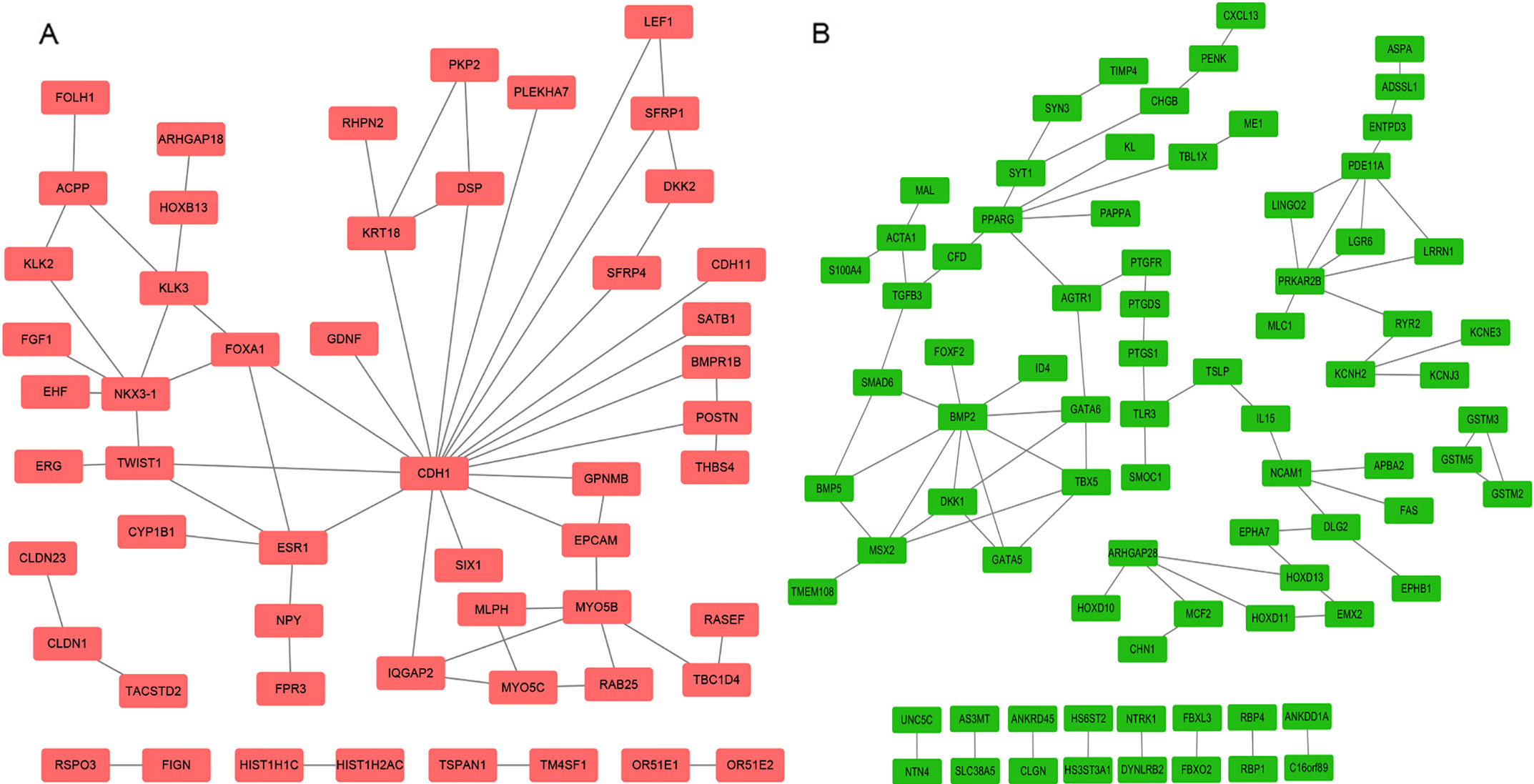
Enriched significant of small molecules
Based on the STRING database, protein network was constructed (Fig. 3). Totally, 198 protein pairs were identified in the network (combined scores of
Identification of small molecular agents
Based on the results of cMap database mapping, a total of 7 small molecular agents were obtained, such as phenoxybenzamine, emetine and fendiline. In Table 2, the small molecular agent phenoxybenzamine had the highest negative score.
Discussion
Prostate cancer is one of the most common urinary tumors and ranks second in death caused by cancer in men in the USA [19, 20]. In current study, we used the gene expression profile obtained from the GEO database, identified 359 DEGs, including 155 upregulated and 204 downregulated genes in PCa. Down-regulated genes were mainly enriched in several functional terms such as cellular response to BMP stimulus, response to BMP, extracellular region and extracellular region part, and pathways associated with TGF-beta signaling pathway. Up-regulated genes were significantly associated with other functional terms, such as cell adhesion, cell-cell junction, cell adhesion molecule binding and protein binding involved in cell-cell adhesion, and were mainly enriched in significant pathways such as cell adhesion molecules (CAMs). In order to clarify the interactions of DEGs, we constructed the PPI network. Five genes were selected as hub genes, including CDH1, NKX3-1, BMP2, PPARG and PRKAR2B. Among the five genes, CDH1 and NKX3-1 were up-regulated, while BMP2, PPARG and PRKAR2B were down-regulated. In addition, small molecular agent phenoxybenzamine was identified as an important potential drug for prostate cancer.
Cell adhesion molecules (CAMs), a group of cell surface receptors, are capable of modulating signal transduction, which is essential for regulation of processes including cellular adhesion, proliferation, differentiation, migration and apoptosis [21, 22]. Study reported that dysregulated CAMs might cause deregulation of these biological processes in tumors, indicating an important role of CAMs in cancer development [23, 24]. Cell adhesion is a common biological process, such as the interaction of cell-cell and cell-matrix [25], involves in cellular growth, differentiation and migration [26].
CDH1 gene is mainly involved in regulating cell adhesion, migration and proliferation of epithelial cells. Mutation of this gene promotes cancer progression by increasing proliferation, invasion, and/or metastasis [27]. The downregulation of CDH1 gene in epithelial cells is usually correlated with tumor formation and differentiation [28]. However, in our study, overexpression CDH1 gene was accordant with GO/KEGG pathway analysis. CDH1 gene expression is higher in prostate cancer than normal samples, which may suggest that overexpression CDH1 plays a minor effect in tumor development. In addition, previous study reported that tumor and normal cells enhanced cell adhesion in the late stage of tumor development [24]. Hence, the expression level of CDH1 in different stages in prostate cancer needs to further study.
TGF-
Bone morphogenetic protein-2 (BMP-2) is a multifunctional growth factor, which strongly induces bone formation and belongs to the TGF-
In addition, NKX3.1 is a homologous domain gene regulated by androgen, which functions mainly as a prostatic tumor suppressor [33]. However, previous studies showed that NKX3.1 protein is positive in primary prostatic adenocarcinomas, downregulated in advanced prostate cancers, and completely lost in metastatic prostate cancers [34, 35]. Furthermore, Xu et al. [36] found NKX3.1 overexpression in 31% tumor specimens, no change in 48% tumor specimens and decreased expression in 21% tumor specimens. In our result, NKX3.1 was an up-regulated gene in primary prostate cancer, suggesting that overexpression of NKX3.1 might lead to tumor development.
PPARG is thought to be a tumor suppressor in various types of tumors including prostatic cancer [37], liposarcoma [38], breast cancer [39], and cervical cancer [40, 41], which induces tumor apoptosis and growth arrest. The lower expression levels of PPARG was reported in cervical adenocarcinoma than matched normal cervical tissue [42]. In our result, PPARG was downregulated in tumor tissue, which is accordance with former studies. Therefore, we further confirm that PPARG might be capable of inhibiting tumor development in PCa. It may be a potential therapeutic biomarker in PCa.
PRKAR2B is a Protein Coding gene, which encodes cAMP-dependent protein kinase type II-beta regulatory subunit in humans [43]. Previous research has demonstrated that PRKAR2B was downregulated in human CCA tissues and CCA cell lines [44]. In addition, our results also revealed PRKAR2B gene was downregulated in prostate cancer. Therefore, PRKAR2B may play an inhibitory role in the development of primary prostate cancer.
At last, the study found candidate small molecules that may be involved in promoting or suppressing the development of PCa. Phenoxybenzamine was considered to be small molecule drug with the highest negative score, which means it maybe suppress tumor progression. It has been reported that Phenoxybenzamine significantly reduced PC3 proliferation, because of the capable to decrease cell proliferation, migration and adhesion [45]. Another molecule, Phenoxybenzamine hydrochloride (PHEN) could inhibit cell growth and invasion in glioma [46], induce cell cycle arrest and apoptosis in prostate cancer cell lines [47]. Additionally, a high-throughput screening study has demonstrated that PHEN was thought to be a novel small molecule inhibitor because of potential anti-cancer effect [48]. Therefore, Phenoxybenzamine or Phenoxybenzamine hydrochloride may be a potential drug to treat PCa.
In conclusion, our study found several significant genes in PCa progression, such as BMP2, PPARG and PRKAR2B, which may have the inhibitory effect in the progression of primary prostate cancer. Furthermore, phenoxybenzamine or Phenoxybenzamine hydrochloride may act as a potential molecular drug to treat PCa. At present, our study may be helpful to elucidate the underlying molecular mechanisms of PCa, and identifies potential candidate targets to treat PCa. However, the current study is performed based on bioinformatics methods and no experiments have been performed to confirm the conclusions. Therefore, further experimental study is required to confirm the findings of the present result.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Acknowledgments
This study was supported by the National Key Research and Development Program of China (2016YFC0902301) and the grants from the Science, Technology and Innovation Committee of Shenzhen Municipality (JSGG20140702161347218 and KJYY20140530141443717).