
Abstract
BACKGROUND:
The runt-related transcription factor family (RUNXs) including RUNX1, RUNX2, and RUNX3 are key transcriptional regulators in normal hematopoiesis. RUNXs dysregulations caused by aberrant expression or mutation are frequently seen in various human cancers especially in acute myeloid leukemia (AML).
OBJECTIVE:
We systemically analyzed the expression of RUNXs and their relationship with clinic-pathological features and prognosis in AML patients.
METHODS:
Expression of RUNXs was analyzed between AML patients and normal controls from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) projects. Correlations between RUNXs expression and clinical features together with survival were further analyzed.
RESULTS:
All RUNXs expression in AML patients was significantly increased as compared with controls. RUNXs expression was found to be significantly associated with genetic abnormalities such as RUNX1 mutation, t(8;21) and inv(16)/t(16;16). By Kaplan-Meier analysis, only RUNX3 overexpression was associated with shorter overall survival (OS) and disease-free survival (DFS) among non-M3 AML patients. Notably, in high RUNX3 expression groups, patients received hematopoietic stem cell transplantation (HSCT) had markedly better OS and DFS than patients without HSCT among both all AML and non-M3 AML. In low RUNX3 expression groups, there were no significant differences in OS and DFS between HSCT and non-HSCT groups among both all AML and non-M3 AML. In addition, a total of 835 differentially expressed genes and 69 differentially expressed microRNAs were identified to be correlated with RUNX3 expression in AML.
CONCLUSION:
RUNXs overexpression was a frequent event in AML, and was closely associated with diverse genetic alterations. Moreover, RUNX3 expression may be associated with clinical outcome, and helpful for guiding treatment choice between HSCT and chemotherapy in AML.
Introduction
The runt-related transcription factor (RUNX) family (RUNXs) including RUNX1, RUNX2, and RUNX3 are key transcriptional regulators in normal hemato- poiesis [1, 2]. RUNXs dysregulations caused by aberrant expression or mutation are frequently seen in various human cancers especially in leukemia [3, 4]. RUNX1/RUNX2/RUNX3 is also called acute myeloid leukemia 1/acute myeloid leukemia 3/acute myeloid leukemia 2 (AML1/AML3/AML2) because of the discovery of its gene sequence from a human patient with acute myeloid leukemia (AML) [5]. RUNX1/AML1 is among the most frequently mutated genes in leukemia and is associated poor prognosis in cytogenetically normal AML (CN-AML) [6]. Moreover, chromosomal translocations involving RUNX1 lead to fusion gene formation, RUNX1-RUNX1T1 being the most common type in several AML subtypes [7]. For RUNX2 and RUNX3, no genetic mutations and translocations are observed among AML patients.
Abnormal expression of RUNXs and their relationship with prognosis have been partly reported in AML. Fu et al. reported that high expression of RUNX1 was associated with unfavorable outcomes in CN-AML [8]. Moreover, high RUNX3 expression was associated with a poor event-free survival among childhood AML [9]. Notably, RUNX3 expression was significantly decreased in AML with the t(8;21) and inv(16)/t(16;16), and probably caused by promoter DNA hypermethylation [9, 10]. To the best of our knowledge, bioinformatics analysis has yet been applied to explore the role of the RUNXs expression in AML. Herein, we systemically analyzed the expression of RUNXs and their relationship with clinic-pathological features and prognosis in patients with AML.
Materials and methods
GEPIA dataset
DNMTs expression in bone marrow cells of AML patients and normal controls (normal bone marrow samples from healthy donors) were analyzed by the Gene Expression Profiling Interactive Analysis (GEPIA) web (
Patients from TCGA
All the 173 de novo AML patients with RUNXs expression data included in this study were obtained from TCGA datasets [12]. The whole-genome sequencing or whole-exome sequencing, along with RNA and microRNA sequencing and DNA-methylation analysis were used to analyze the gene expression, mutation and methylation in these de novo AML patients [12]. Clinical and molecular characteristics were obtained, including, age, sex, white blood cell (WBC) counts, peripheral blood (PB) blasts, bone marrow (BM) blasts, French-American-British (FAB) subtypes, and the frequencies of genetic mutations as presented in Table 1. After induction chemotherapy for all 173 AML patients, consolidation treatment included chemotherapy for 100 patients and hematopoietic stem cell transplantation (HSCT) for 73 patients.
Bioinformatics analyses
The details for the identification of microRNAs targeting RUNX3 were reported as our previous study [13, 14].
Statistical analyses
SPSS 22.0 were used for statistical analyses and figures creation. Mann-Whitney’s U test was used for the comparison of continuous variables, whereas Pearson Chi-square analysis or Fisher exact test was applied for the comparison of categorical variables. The prognostic effect of RUNXs expression was evaluated using disease-free survival (DFS) and overall survival (OS) analyzed though Kaplan-Meier analysis. The two-tailed
Correlation of RUNXs expression with clinic-pathologic characteristics in AML
Correlation of RUNXs expression with clinic-pathologic characteristics in AML
RUNX family expression in AML. RUNX1/2/3 expression in controls from GTEx projects and AML patients from TCGA datasets using the GEPIA website (
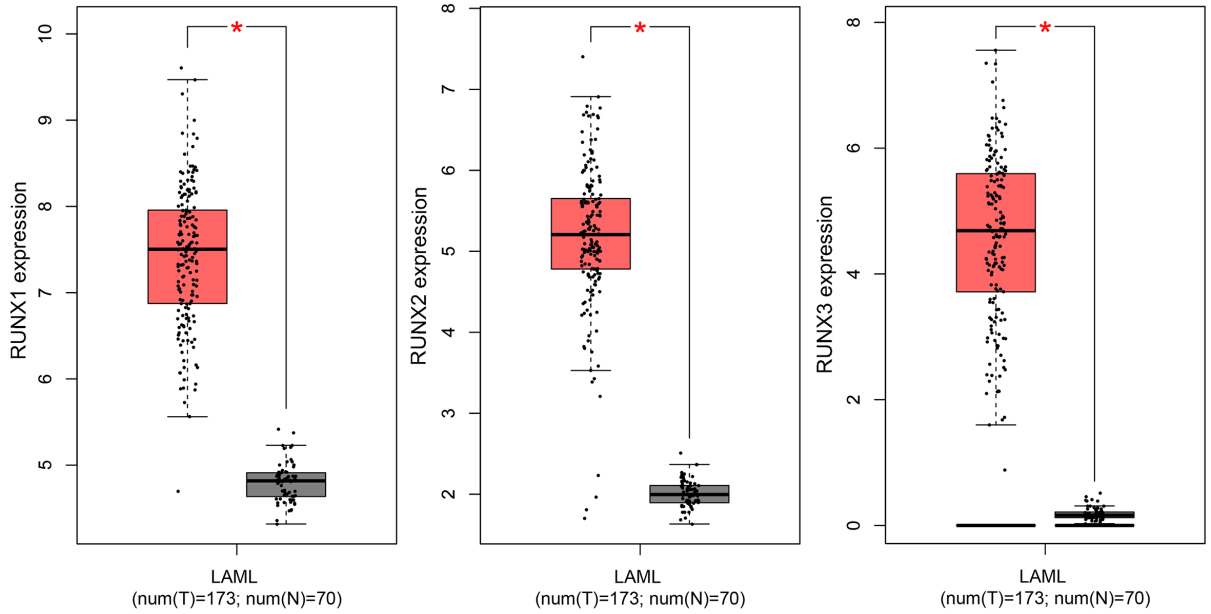
RUNXs expression in AML
Using the online web GEPIA (
Clinical characteristics of AML patients with aberrant RUNXs expression
To explore the associations of RUNX1/2/3 expression with clinical characteristics in AML, we further divided these AML patients into two groups based on median level of RUNX1/2/3 expression, respectively. The comparison of clinical characteristics of the AML patients between two groups was summarized in Table 1. High RUNX1 expression was significantly associated with higher peripheral blood (PB) blasts (
RUNX family expression in AML patients with and without common gene mutations. 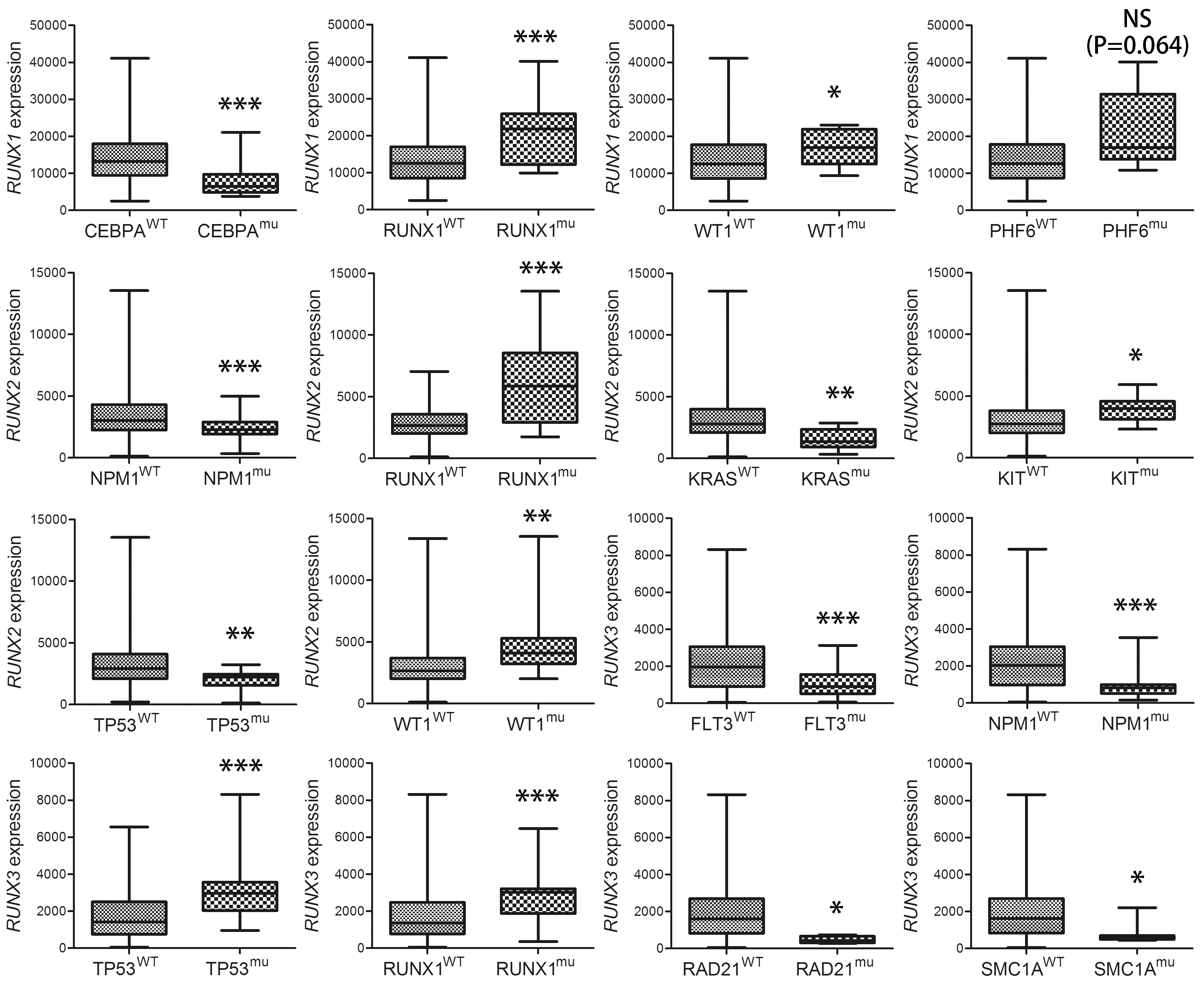
We further observed the association of RUNXs expression with gene mutations in AML. Although there were less mutational cases (
Regarding the significant associations, we further compared the RUNX1/2/3 expression level between two groups with and without these gene mutations, which were presented Fig. 2.
The effect of abnormal RUNXs expression on survival in AML
We next evaluate the prognostic effect of RUNXs expression on survival. By Kaplan-Meier analysis, patients with high RUNX1 expression showed nearly same overall survival (OS) and disease-free survival (DFS) time compared with those with low RUNX1 expression among both whole-cohort and non-M3 AML (Fig. 3a,
The impact of aberrant RUNX family expression on survival of AML patients. (a) Overall survival (OS) and disease-free survival (DFS) between low and high RUNX1 expression among both whole-cohort and non-M3 AML analyzed by Kaplan-Meier survival. (b) OS and DFS between low and high RUNX2 expression among both whole-cohort and non-M3 AML analyzed by Kaplan-Meier survival. (c) OS and DFS between low and high RUNX3 expression among both whole-cohort and non-M3 AML analyzed by Kaplan-Meier survival.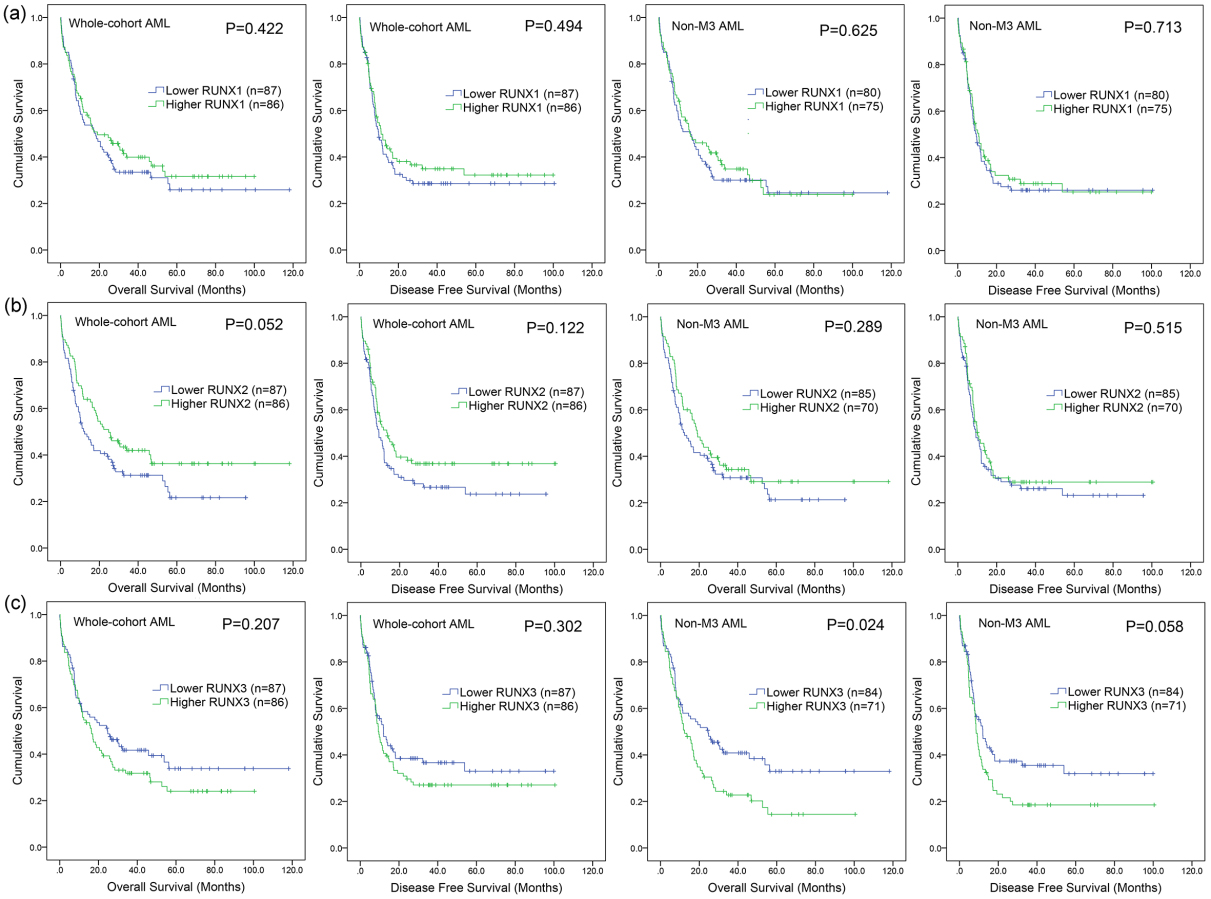
Because RUNX3 overexpression predicted poor clinical outcome in AML, we intended to investigate whether patients with RUNX3 overexpression could benefit from hematopoietic stem cell transplantation (HSCT). We compared OS and DFS between patients with and without HSCT among both high and low RUNX3 expression groups. In high RUNX3 expression groups, patients received HSCT had markedly longer OS and DFS than patients without HSCT among both all AML (Fig. 4,
The effect of HSCT on survival of AML patients among high and low RUNX3 expression groups. Overall survival (OS) and disease-free survival (DFS) between patients with and without HSCT among both high and low RUNX3 expression groups analyzed by Kaplan-Meier survival.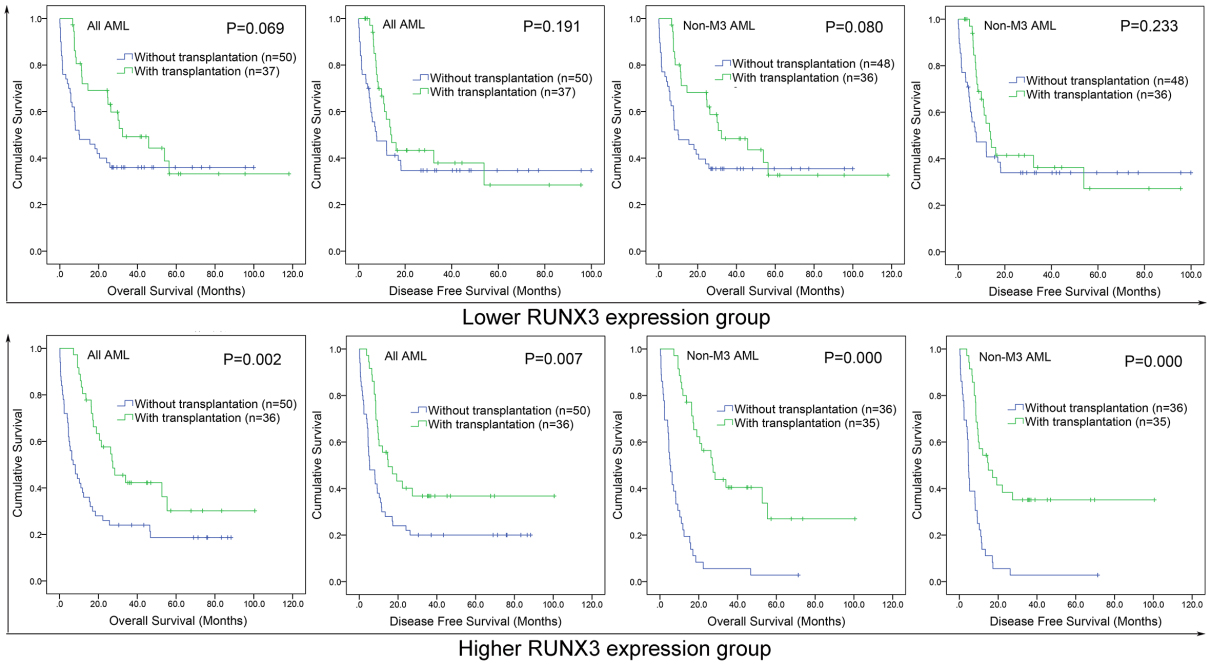
To gain insights into the biological function of RUNX3 in AML, we first compared the transcriptomes of high and low RUNX3 expression groups. A total of 835 differentially expressed genes were identified including 653 positively correlated genes and 182 negatively correlated genes (FDR
Molecular signatures associated with RUNX3 in AML. (A) Expression heatmap of differentially expressed genes between AML patients with high and low RUNX3 expression groups (FDR 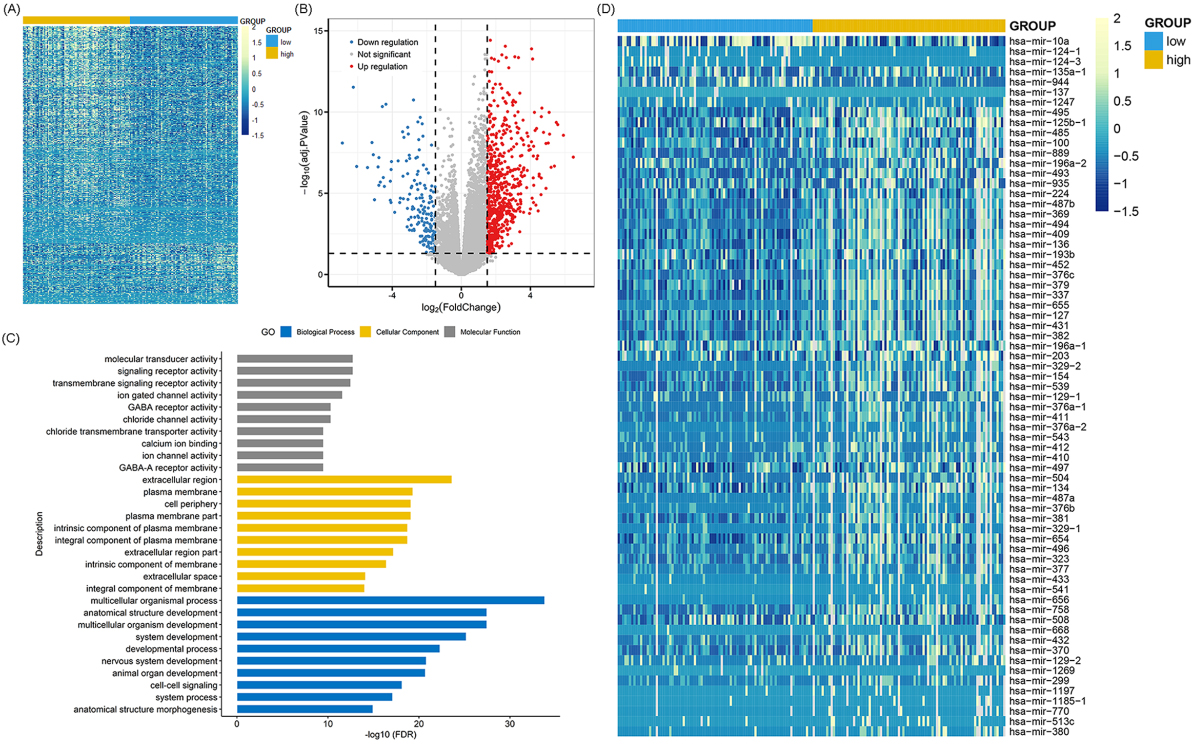
We also compared microRNAs expression between high and low RUNX3 expression groups. A total of 69 differentially expressed microRNAs were identified including 62 positively correlated genes and 7 negatively correlated genes (FDR
In this study, we showed that RUNX1/2/3 expression was significantly increased in AML, suggesting that RUNXs played crucial roles in leukemogenesis. The potential role of RUNXs in AML was preliminarily investigated. Although the earliest study suggested that RUNX1 acted as a tumor suppressor gene in AML [22], it was now understood that RUNX1 functions as an oncogene necessary to sustain AML [23, 24, 25, 26]. For RUNX2, although little studies showed its direct function in AML, a number of investigations showed the oncogenic role of RUNX2 in various human solid tumors [27]. However, the potential role of RUNX3 remains controversial in human cancers [28]. Several studies reported the anti-cancer effects of RUNX3 in several cancers such as colorectal cancer, lung cancer, glioma and gastric cancer [29, 30, 31, 32]. However, Chen et al. showed that RUNX3 promoted the tumorigenic phenotype in a human granulosa cell tumor-derived cell line KGN [33]. In addition, the oncogenic role of RUNX3 was also observed in NK/T-cell lymphoma, basal cell carcinomas, head and neck cancer, ovarian cancer, Ewing sarcoma and oral squamous cell carcinoma [34, 35, 36, 37, 38, 39]. By our study, the interactions of RUNX3 with several microRNAs including miR-137, miR-124, miR-125b, and miR-100 also supported the oncogenic role of RUNX3 in AML [18, 19, 20, 21]. Similarly, several microRNAs were also reported to be interacted with RUNX1 in normal and malignant myelopoiesis [40]. These results suggested that RUNX3 functioned as different roles among different cancer type or genetic context.
We next showed that aberrant RUNXs expression was closely associated with specific cytogenetic abnormalities and molecular gene alterations in the patients with AML. Previously, Fu et al. presented that RUNX1 overexpression was associated with FAB-M2, FLT3-ITD mutations, and CEBPA wild-type [8]. Although we also observed the significant associations of RUNX1 overexpression with CEBPA wild-type, we did not find the associations of RUNX1 expression with other molecular abnormalities. These may be caused by the limited samples and the heterogeneous cohorts of AML patients from different ethnicities. Accordingly, small cases of the mutational samples also urged us to confirm our results. In addition, RUNX1 mutations were also associated with distinct clinico-pathologic and genetic features in AML [41, 42]. Functional studies showed that the role of RUNX1 in AML often cooperated with cytogenetic and molecular abnormalities such as FLT3-ITD and t(4;11)/MLL-AF4 [26, 43]. For RUNX3, several studies have reported the significant relations of RUNX3 underexpression or hypermethylation with t(8;21) and inv(16)/t(16;16) translocations [9, 10]. Herein, we also observed the phenomena in accordance with the previous studies. However, due to the few studies focused on the correlation of RUNXs expression with specific cytogenetic and molecular abnormalities, the significant associations of RUNX2/RUNX3 overexpression with other molecular alterations needs further studies. In spite of this fact, all these results further support the hypothesis that the role of RUNXs has been involved in diverse signaling pathways and cellular processes may through multiple protein-interacting partners.
Because AML is a highly heterogeneous myeloid malignancy, its treatment needs to be more personalized and precise, and is mainly based on the risk classifications [44]. AML patients with high risks surely need intensive therapy especially HSCT to improve survival [45]. Consequently, identification of novel biological markers which could predict outcome or guide treatment choice will make more contributions to the clinical management of newly diagnosed AML. Herein, we further revealed that RUNX3 overexpression was associated with poor clinical outcome in AML. In accordance with our studies, a recent study also showed the negative effect of RUNX3 overexpression on prognosis in AML [46]. Moreover, RUNX3 expression pattern may serve as a decision biomarker which could guide treatment choice between HSCT and chemotherapy in AML after induction therapy. However, it is the first study reported RUNX3 expression as a potential prognostic and predictive biomarker in AML. Therefore, randomized prospective trials and basic mechanism studies are needed to confirm and expand our results before RUNX3 expression pattern can be used routinely as a potential prognostic biomarker guiding treatment choice for newly diagnosed AML.
Collectively, RUNXs overexpression was a frequent event in AML, and was closely associated with diverse genetic alterations. Moreover, RUNX3 expression may be associated with clinical outcome, and helpful for guiding treatment choice between HSCT and chemotherapy in AML.
Footnotes
Acknowledgments
The work was supported by National Natural Science foundation of China (81900166), Zhenjiang Clinical Research Center of Hematology (SS2018009).
Conflict of interest
The authors declare that they have no competing interests.