
Abstract
BACKGROUND:
Lung adenocarcinoma is the most common type of lung cancer, and it is one of the most aggressive and rapidly fatal tumor types.
OBJECTIVE:
To identify a signature mutation genes for prognostic prediction of lung adenocarcinoma.
METHODS:
Four hundred and sixty-two lung adenocarcinoma cases were screened out and downloaded from TCGA database. Mutation data of 18 targeted genes were detected by MuTect. LASSO-COX model was used to screen gene loci, and then a prognosis model was established. Afterwards, 40 clinical patients of lung adenocarcinoma were collected to verify the mutation features and the predictive function of the above prognostic model. The mutations of above 18 genes were sequenced with targeted next generation sequencing (NGS) and analyzed with GATK and MuTect.
RESULTS:
TP53 (282, 32.38%), NF1 (82, 9.41%) and EGFR (80, 9.18%) were the top 3 most frequent mutation genes. A total of 7 variables were screened out after lasso-COX analysis (tumor stage, age, diagnostic type, SMARCA4, GNAS, PTCH2, TSC2). SMARCA4, GNAS and TSC2 were a gene mutation signature to predict a poor prognosis.
CONCLUSIONS:
We established a prognostic model for lung adenocarcinoma, and further concluded that SMARCA4, GNAS and TSC2 were a gene signature which plays a prognostic role.
Introduction
Lung cancer is “the double most cancer” with the highest morbidity and the leading cancer-associated mortality in the worldwide. It is estimated that there are 228,150 new cases of lung cancer and 142,670 deaths by the American Cancer Society in 2019 [1]. Lung adenocarcinoma is the most common type of lung cancer, comprising around 40% of all lung cancer cases, and it is one of the most aggressive and rapidly fatal tumor types with overall survival less than 5 years [2]. Outcome of patients with lung adenocarcinoma treated with chemotherapy is usually poor, and it is improved with the development of targeted therapy and immunotherapy [3, 4]. However, the prognosis of patients with lung adenocarcinoma is still depressing with a 5-year survival of 5–16% [5]. Predicting prognosis is helpful to formulate reasonable follow-up treatment plan and reduce ineffective or over-treatment. The gene mutations have been studied as the prognosis prediction markers for lung adenocarcinoma. In Asia-Pacific region, EGFR exon 18/21 mutation is the most common type of oncogenic mutation in lung adenocarcinoma, with incidence rate of 55% [6]. KRAS and TP53 have also been shown to be a negative prognostic marker for lung adenocarcinoma [7]. PIK3CA gene mutation associated with poor prognosis of lung adenocarcinoma [8]. Nevertheless, most gene mutations have a more clear predictive effect on target therapy, and have a poor predictive effect, or sometimes wrong, on chemotherapy, radiotherapy and various combination therapies [9, 10]. There is still a lack of effective prognostic judgement and evaluation index of chemotherapy efficacy in clinic. Additionally, due to individual differences and the cost of gene testing, mutation genes that are really useful for clinical use are very limited. Therefore, it is necessary and meaningful to find more oncogenic mutations and their role in predicting the prognosis.
In this article, the prognostic correlation of 18 genes related to lung cancer was analyzed, namely, TP53, EGFR, SMARCA4, HGF, ERBB4, PDGFRB, NF1, GNAS, C11orf30, BRCA2, PTCH2, DDR2, MAPK3, TSC2, FLT4, APC, SMO and ABL1. The aim was to identify a signature mutation genes for prognostic prediction of lung adenocarcinoma on the basis of further understanding of its molecular genetic characteristics.
Materials and methods
TCGA data screening
Tissue mutation data were downloaded from The Cancer Genome Atlas (TCGA), and complete clinical data sets are available at the TCGA website (
Patients and samples
Forty patients diagnosed as lung adenocarcinoma were collected from February 2013 to April 2019 in Tianjin Chest Hospital. The tumor tissues were sequenced with targeted next generation sequencing (NGS) of 18 lung cancer gene panel, and the paracancerous tissues or leukocyte were as controls.
Sequencing (targeted NGS)
Fresh tissues samples were used for DNA extraction with Genomic DNA extraction kit (Qiagen, Valencia, CA) strictly following the manufacturer’ s instructions. The DNA samples were purified by Agencourt AMPure XP beads (Agentcourt Biosciences, Beverly, MA). Afterwards, the adaptor library was amplified and linked, and the total library was accurately quantified by Qubit DNA HS Assay Kit (Invitrogen, USA, CA). A library hybridization kit, SeqCap EZ MedExome Enrichment kits (Roche, Basel, CH), was used to capture target sequences and bead capture and elution hybridization libraries with Roche’s customized 18 targeted gene probes (Roche, Basel, CH). After amplifying the captured library by PCR, the constructed library was sequenced by Illumina MiniSeq sequencer. The average sequencing depth of tissue samples was 500 X. It could detect mutations with very low frequency to 0.1%.
Mutation analysis
Using SOAPnuke to filter the sequencing data, removing the sequence containing adapter and the low quality data, and the quality of the original data after filtering meets Q30 85%. BWA was used to compare data to the Human Reference Genome (hg19.fa); Genome Analysis Toolkit (GATK) version 2.3.9 was adapted to re-compare the reads in the interval, calibrate and rearrange the alkali matrix quality values, and to count the sequence depth and coverage. Insertion/deletion (Indel) and single nucleotide polymorphism (SNP) were detected by MuTect version 2.0. SNP and Indel with more than 1% allele mutation frequencies were filtered out. Furthermore, the functions of mutations were annotated with PolyPhen version 2, a software to predict the possible impact of an amino acid substitution on the structure and function of a human protein. The higher the score, the greater the hazard. The classification criteria: probably damaging 0.90–1.0, possibly damaging 0.45–0.90, benign 0–0.45.
Clinical characteristics of TCGA cases
Clinical characteristics of TCGA cases
TCGA, The Cancer Genome Atlas.
The statistical analysis was performed using IBM SPSS statistics 21 and Rstudio R3.4.4 software. The Chi-square test was used to determine the correlation between gene mutations and clinical indicators, and the fisher test was used when the sample size was too small. Establishing a prognostic model using the Lasso model and COX model, and the proportional hazards (PH) assumption in the COX model is assumed to use the Schoenfeld residual test. Besides, the prognosis model was evaluated and validated by K-M survival analysis, ROC curve and clinical validation data.
Results
Clinical characteristics
The clinical characteristics of 462 cases were listed in Table 1. In the cohort, 55.6% were female 44.4% male, and 79% were white race, and 78.5% were I–II stage.
Mutation features and Clinical indicator correlation
In the 462 cases, a total of 871 mutations were screened out, namely, TP53 (282, 32.38%), NF1 (82, 9.41%), EGFR (80, 9.18%), ERBB4 (63, 7.23%), HGF (58, 6.66%), SMARCA4 (50, 5.74%), BRCA2 (39, 4.48%), APC (35, 4.02%), FLT4 (30, 3.44%), DDR2 (27, 3.10%), GNAS (25, 2.87%), C11orf30 (21, 2.41%), PDGFRB (19, 2.18%), ABL1 (17, 1.95%), TSC2 (16, 1.84%), SMO (15, 1.72%), MAPK3 (7, 0.80%) and PTCH2 (5, 0.57%). The mutation features of them were showed in Fig. 1A–C. Among them, 768 were SNPs (88.2%), 84 deletions (9.6%), and 19 inserts (2.2%) (Fig. 1A); In the 768 SNPs, 24.74% were C-A (
The mutation features of targeted genes. A. The overall catastrophe type distribution; B. The location and possible functional significance of mutations, None indicated the functional significance of mutations were unknown or unclear in Polyphen; C. The mutation forms of bases of SNPs.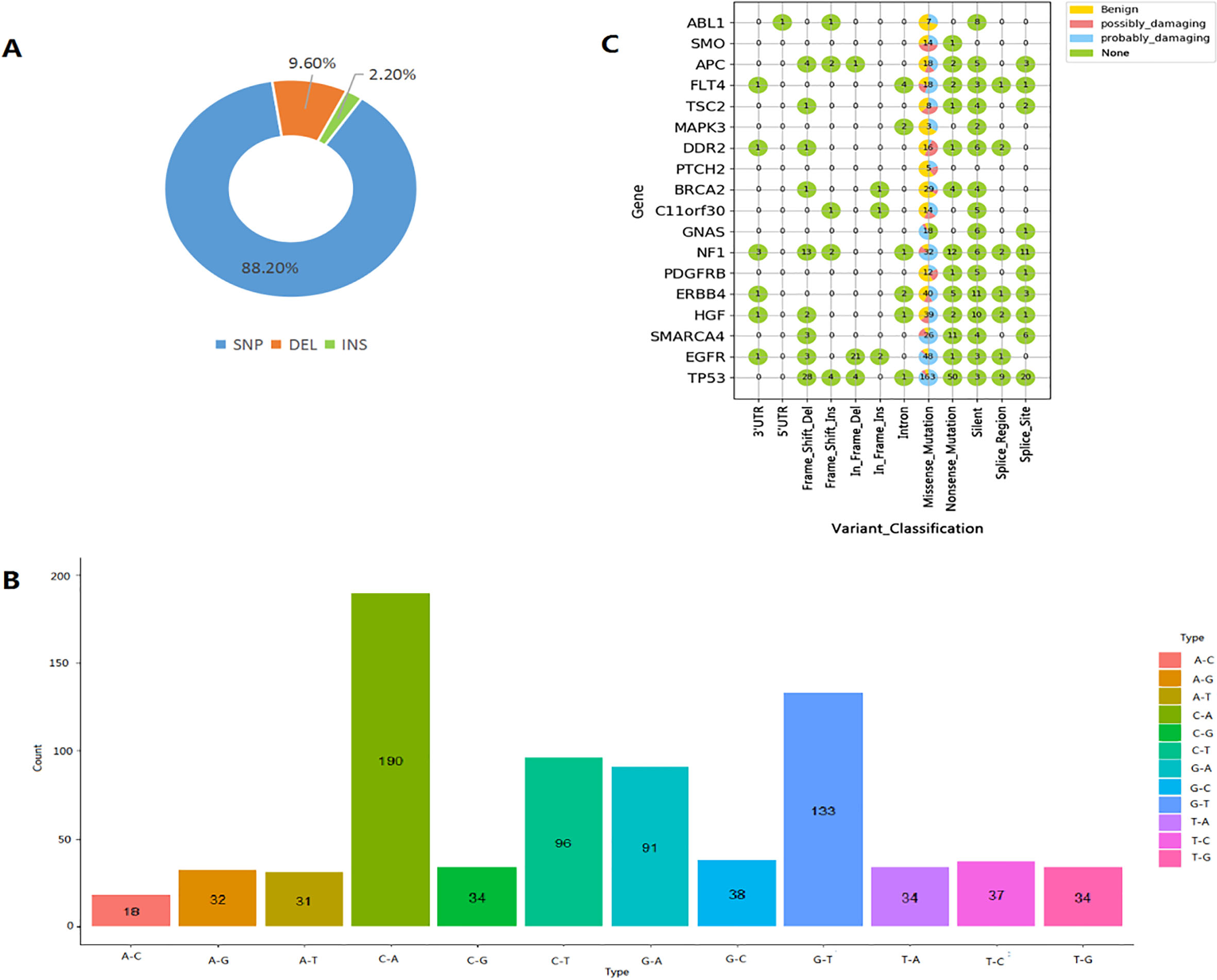
The prognostic model. A. COX variable selection cyclic voltammograms based on LASSO method; B. The schoenfeld Residual graph of the 6 screened variables, When the 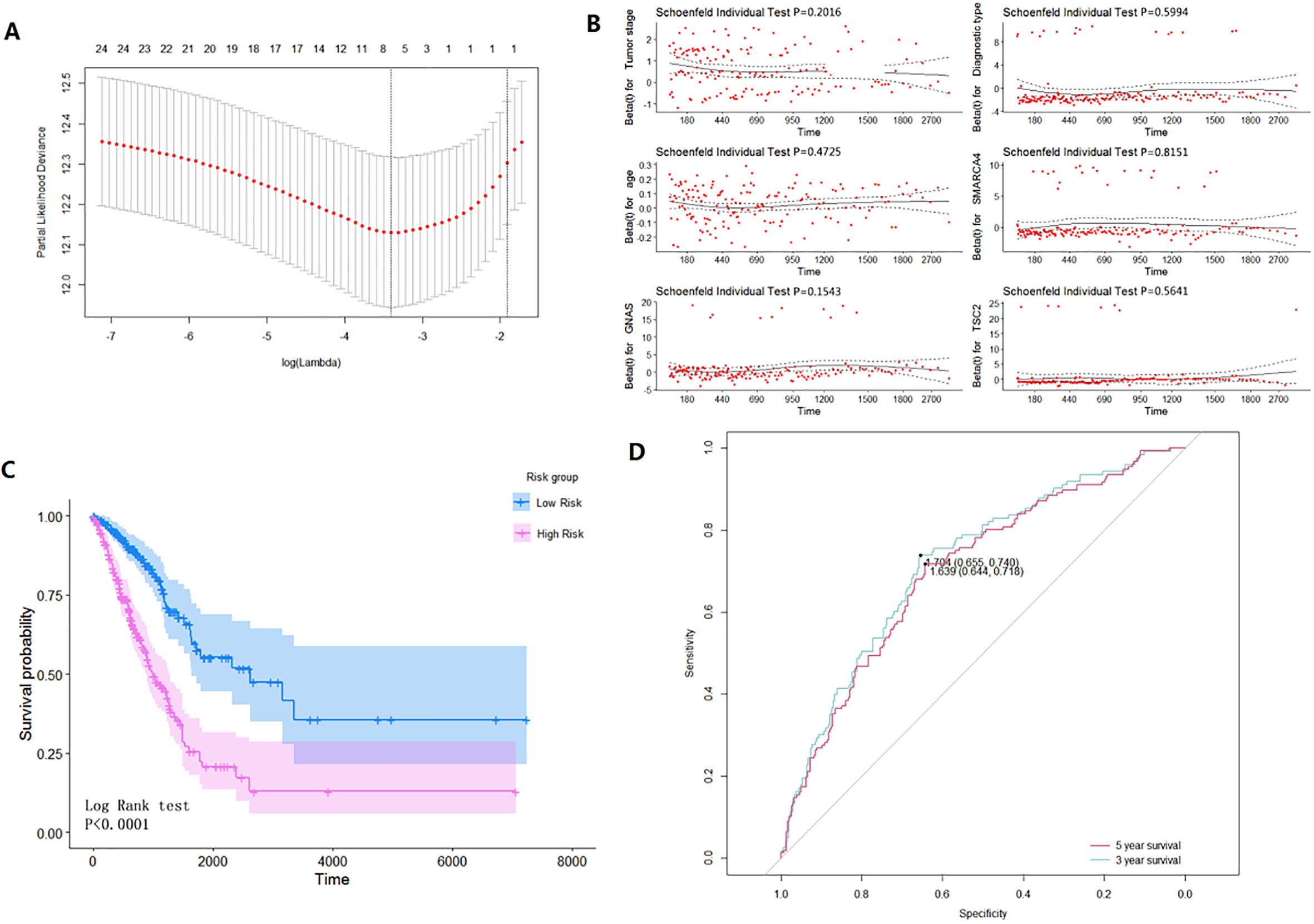
The verification results in clinical cases. A. Comparison of the mutation frequencies of all 18 targeted genes between TCGA and clinical cases; B. Comparison of the mutation frequencies of 18 targeted genes between TCGA and clinical cases in subgroups of stage I–II and stage III–IV; C. The number of mutations in each gene in 40 clinical cases, the size of dots indicates the number of mutations, and the color of dots represents different genes; D. The overall survival curves of low-risk and high-risk groups based on above prognosis model.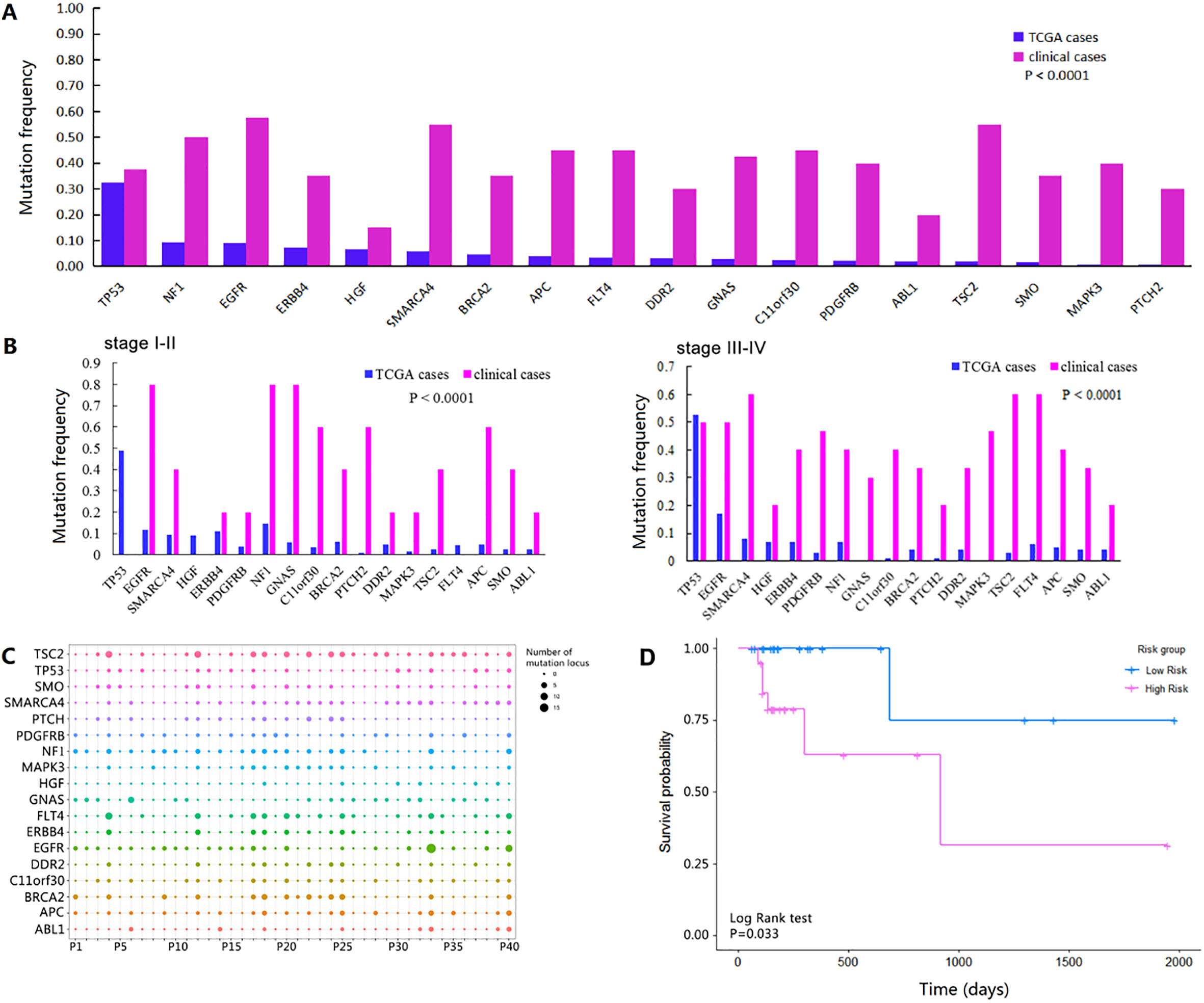
As for the possible impact on the structure and function of a human protein, 121 were benign, 62 possibly damaging, and 316 probably damaging. Additionally, all of them, with relatively clear functions, were missense mutations.
We used the lasso model to select meaningful variables. The lasso model introduced 23 variables, including gender, tumor stage, tumor location, age, diagnosis type, and 18 gene mutation information. After cross-validation, the best lambda value, lambdamin
Clinical verification
Forty clinical patients of lung adenocarcinoma were collected to verify the mutation features and the predictive function of the above prognostic model with age of 63.75
Discussion
The combined application of multiple marker genes can improve the accuracy of lung cancer screening and diagnosis. The combined application of multiple marker genes and multiple targets can reduce the probability of false positive or false negative, thereby improving the diagnostic efficiency of gene detection [11]. The current study used multiple mutation genes to predict the prognosis of patients with lung adenocarcinoma. LASSO was first proposed by Robert Tibshirani in 1996, which added a Lp norm as a penalty constraint in the calculation of really simple syndication (RSS) minimization [12]. When the lambda component is large, the Lp norm can reduce the coefficients of some parameters with smaller coefficients to 0. By choosing the absolute value of compression coefficient and adjusting the parameter lambda, LASSO can realize the continuous stable process of variable selection and estimation at the same time, so it is widely used. The LASSO method can remove some meaningless or insignificant variable coefficients by adding a constraint condition to the absolute value of the coefficients, so that a better model can be obtained. COX proportional risk regression model is a model with survival outcome and survival time as dependent variables, which is mostly used in the correlation analysis of patient survival data. Combining the characteristics of the two models, we analyzed the prognostic risk of 462 TCGA patients by LASSO-COX model. Subsequently, SMARCA4, GNAS and TSC2 were excavated to risk factors with poor outcome (Fig. 2). In addition, it was further validation in 40 clinical cases (Fig. 3D).
The encoded protein of SMARCA4 is part of the large ATP-dependent chromatin remodeling complex SNF/SWI, which is required for transcriptional activation of genes normally repressed by chromatin [13]. In addition, this protein can bind BRCA1, as well as regulate the expression of the tumorigenic protein CD44 [14]. Some studies have indicated that the high expression of SMARCA4 was associated with the dismal prognosis of lung adenocarcinomas [15, 16, 17]. The present study demonstrated from the risk index formula that the mutation of SMARCA4 was a poor outcome risk for lung adenocarcinomas, which was similar with previous studies. We have not studied the specific mechanism of action, and the relevant report holds that BRG1/SMARCA4 inactivation promotes NSCLC aggressiveness by altering chromatin organization [18]. GNAS, also known as GNAS1, mainly encodes a stimulatory alpha subunit of guanine nucleotide binding protein (G protein), which activates adenylate cyclase in G protein-coupled receptor signaling pathway, leading to an increase in the level of cAMP, and then participates in the regulation of cell growth and cell division [19]. At present, GNAS1 point mutation T393C is widely studied, which is a predictor of recurrence and survival of resectable NSCLC, particularly the advanced NSCLC, and a predictor of chemosensitivity and survival rate of advanced NSCLC patients treated with gemcitabine combined with platinum [20, 21, 22]. Our study found that GANS mutation is a poor prognostic factor in lung adenocarcinoma patients, consistent with previous studies. TSC2 associates with hamartin in a cytosolic complex, possibly acting as a chaperone for hamartin, and the protein Alternative splicing results in multiple transcript variants encoding different isoforms [23, 24]. Mutations of TSC2 lead to tuberous sclerosis complex and its gene product is believed to be a tumor suppressor and is able to stimulate specific GTPases [25, 26]. There are few studies on TSC2 in the prognosis of lung adenocarcinoma. Our results suggested for the first time that TSC2 mutation might be a poor prognostic factor of lung adenocarcinoma. However, this result needs further confirmation. Concurrently, we speculated that SMARCA4, GNAS and TSC2 were a gene mutation signature to predict a poor prognosis. In the future, we would further study the accuracy and specificity of the prognosis of three genome combinations and one or two genes alone, and further explore the relevant molecular mechanisms.
In summary, we established a prognostic model for lung adenocarcinoma, and further concluded that SMARCA4, GNAS and TSC2 were a gene signature which plays a prognostic role.
Footnotes
Acknowledgments
This work was supported by the 2011 Key Project of Medical Science Research of Health Commision of Hebei Province (No. 20110444).
Conflict of interest
The authors declare no conflicts of interest.