Abstract
BACKGROUND:
Lung squamous cell carcinoma (LUSC) is malignant disease with poor therapeutic response and unfavourable prognosis.
OBJECTIVE:
This study aims to develop a long non-coding RNA (lncRNA) signature for survival prediction in patients with LUSC.
METHODS:
We obtained lncRNA expression profiles of 493 LUSC cases from The Cancer Genome Atlas, and randomly divided the samples into a training set (
RESULTS:
A lncRNA-focused risk score model was then constructed for prognosis prediction in the training set and further validated in the testing set and the entire set. Finally, bioinformatics analysis was carried out to explore the potential signaling pathways associated with the prognostic lncRNAs. A set of 9 lncRNAs were found to be strongly correlated with overall survival of LUSC patients. These 9 lncRNAs were integrated into a prognostic signature, which could separate patients into high- and low-risk groups with significantly different survival times in the training set (median: 30.5 vs. 80.5 months, log-rank
CONCLUSIONS:
Our study demonstrated the potential clinical implication of the 9-lncRNA signature for survival prediction of LUSC patients.
Introduction
Lung cancer is one of the most commonly diagnosed cancers around the world, contributing to over 18% of all cancer-related deaths in both male and female [1]. The major pathological type of lung cancer was non-small cell lung cancer, which mainly includes lung adenocarcinoma and lung squamous cell carcinoma (LUSC). Patients with LUSC are often diagnosed in advanced stage when currently available therapy cannot be timely administered [2]. To the treatment by chemotherapy or radiotherapy, LUSC patients are also not as sensitive as small cell cancer patients. In addition, the prognosis of LUSC is poor, with an estimated 5-year survival rate of
It is generally accepted that protein-coding genes (mRNAs) only constitute a small fraction of human genome, while more than 98% of the genome may be processed into non-coding RNAs (ncRNAs) [4]. Long non-coding RNAs (lncRNAs) are a newly discovered major subtype of ncRNAs with
In this study, by integrating lncRNA expression profiles and related clinical information of LUSC patients from The Cancer Genome Atlas database (TCGA,
Materials and methods
LUSC patient data
The level-3 RNA sequencing data (HTSeq-FPKM) and relevant clinical details of TCGA-LUSC cohort were downloaded from TCGA database. After removal of LUSC samples with incomplete survival data and normal samples, a total of 493 patients with LUSC were enrolled in this study. LncRNAs and mRNAs were recognized through mapping gene identification to the annotations from the GENCODE (
Clinical characteristics of patients with LUSC
Clinical characteristics of patients with LUSC
LUSC, lung squamous cell carcinoma.
The association between lncRNA expression and patient’s OS was evaluated using univariate Cox regression analysis. With a parametric test (
The nine lncRNAs significantly associated with overall survival in the training set
The nine lncRNAs significantly associated with overall survival in the training set
Random survival forest algorithm for lncRNA selection. A, error rate for the data as a function of trees; B, out-of-bag importance values for lncRNA predictors.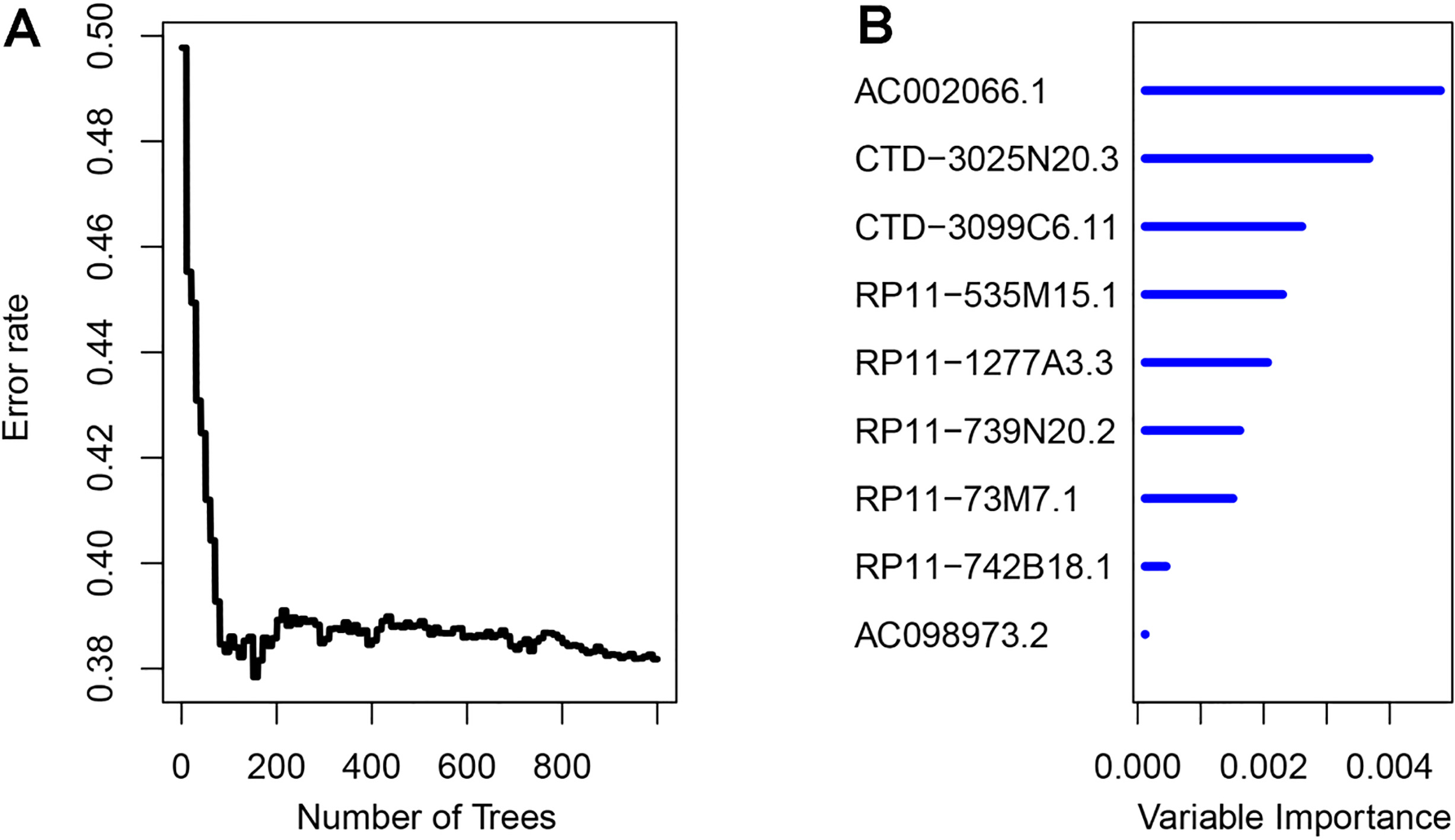
Identification and evaluation of the 9-lncRNA signature for OS prediction of LUSC patients. A, the five-year ROC curve for OS prediction by the 9-lncRNA signature in the training set. B–D, Kaplan-Meier analysis for OS prediction by the 9-lncRNA signature in the training, the testing, and the entire sets.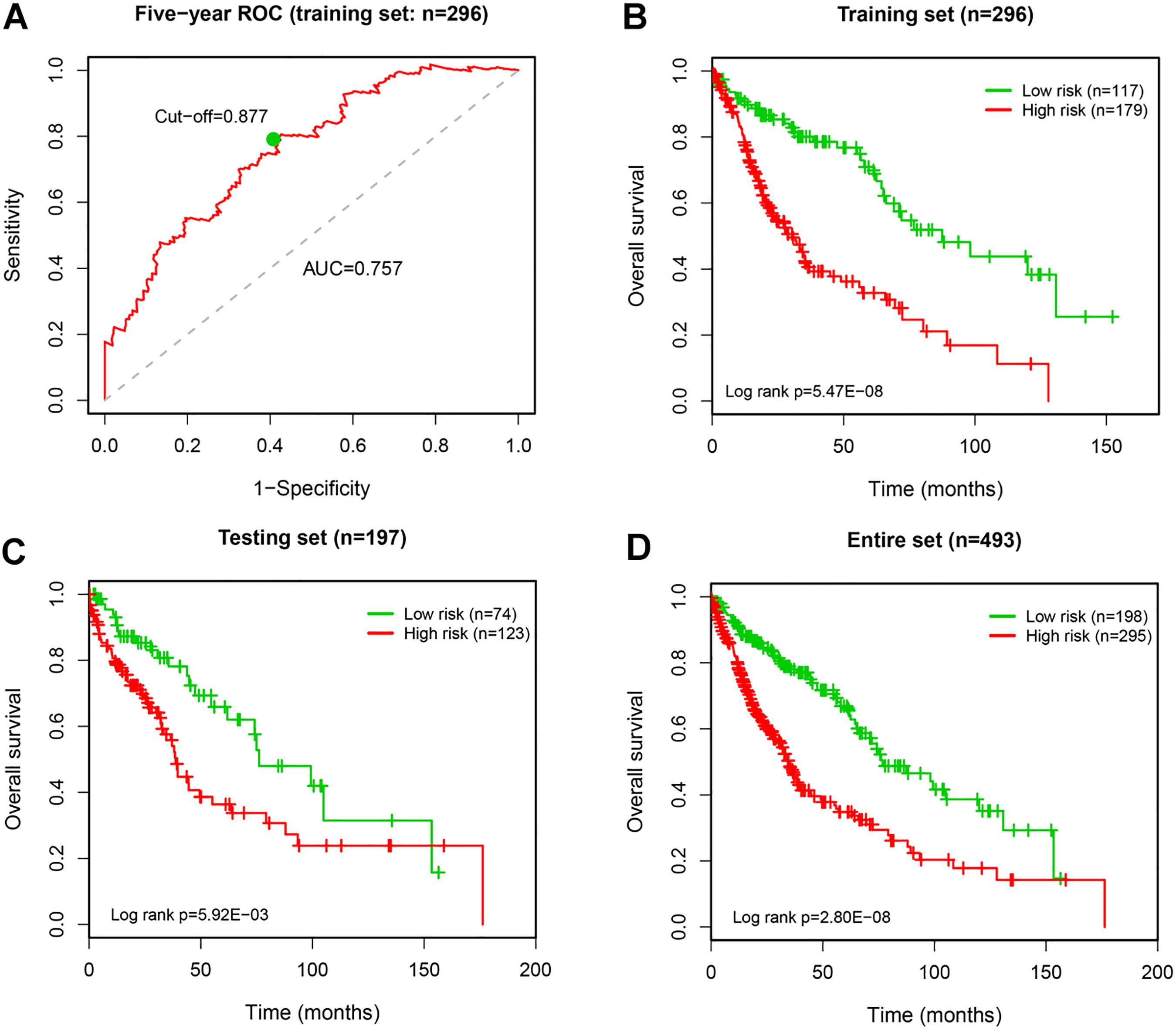
To assess the prognostic performance of the lncRNA risk score signature, we conducted time-dependent receiver operating characteristic (ROC) analyses using R “survivalROC” package [19]. LUSC Patients were then assigned into high- and low-risk groups based on the optimal cut-off value of risk score from the 5-year ROC curve. Survival analysis was performed using Kaplan-Meier method, and log-rank test was used to evaluate the OS difference between high- and low-risk groups. Univariate and multivariate Cox regression analyses were implemented to examine whether the prognostic value of lncRNA signature was independent of clinical features. The statistical significance was based on
Functional enrichment analysis
Pearson correlation coefficients were calculated to identify the co-expression relationship between lncRNAs and mRNAs. To reduce the potential of false positives, we selected only the lncRNA-mRNA pairs with correlation coefficients greater than 0.35. Kyoto encyclopedia of genes and genomes (KEGG) pathway for the mRNAs co-expressed with the prognostic lncRNAs were explored using KOBAS web server (version 3.0,
Results
Identification of a prognostic lncRNA signature in the training set
A flowchart for data processing and analysis is shown in Supplementary Fig. 1. By using univariate Cox regression, we identified a total of 43 candidate lncRNAs that were strongly correlated with patients’ OS in the training set. Based on the RSFVH algorithm, 9 lncRNAs were further selected as prognostic biomarkers. Table 2 showed the genomic details of these 9 lncRNAs with their statistics from univariate Cox regression. In Fig. 1, we can see that AC002066.1 had the greatest importance value than other predictors. Besides, the positive coefficients of 5 lncRNAs indicated that their higher expressions were correlated with shorter survival, and the negative coefficients of the other 4 lncRNAs suggested that their higher expressions were associated with longer survival. By linear combination of the expression levels of the 9 lncRNAs weighted by their Cox regression coefficients, a 9-lncRNA risk-score signature was constructed as follows: risk score
Univariate and multivariate Cox regression analysis for overall survival
Univariate and multivariate Cox regression analysis for overall survival
CI, confidence interval; HR, hazard ratio.
Kaplan-Meier analysis for OS prediction of LUSC using the 9-lncRNA signature in the subanalyses by age (A and B), smoking status (C and D), and tumor stage (E and F).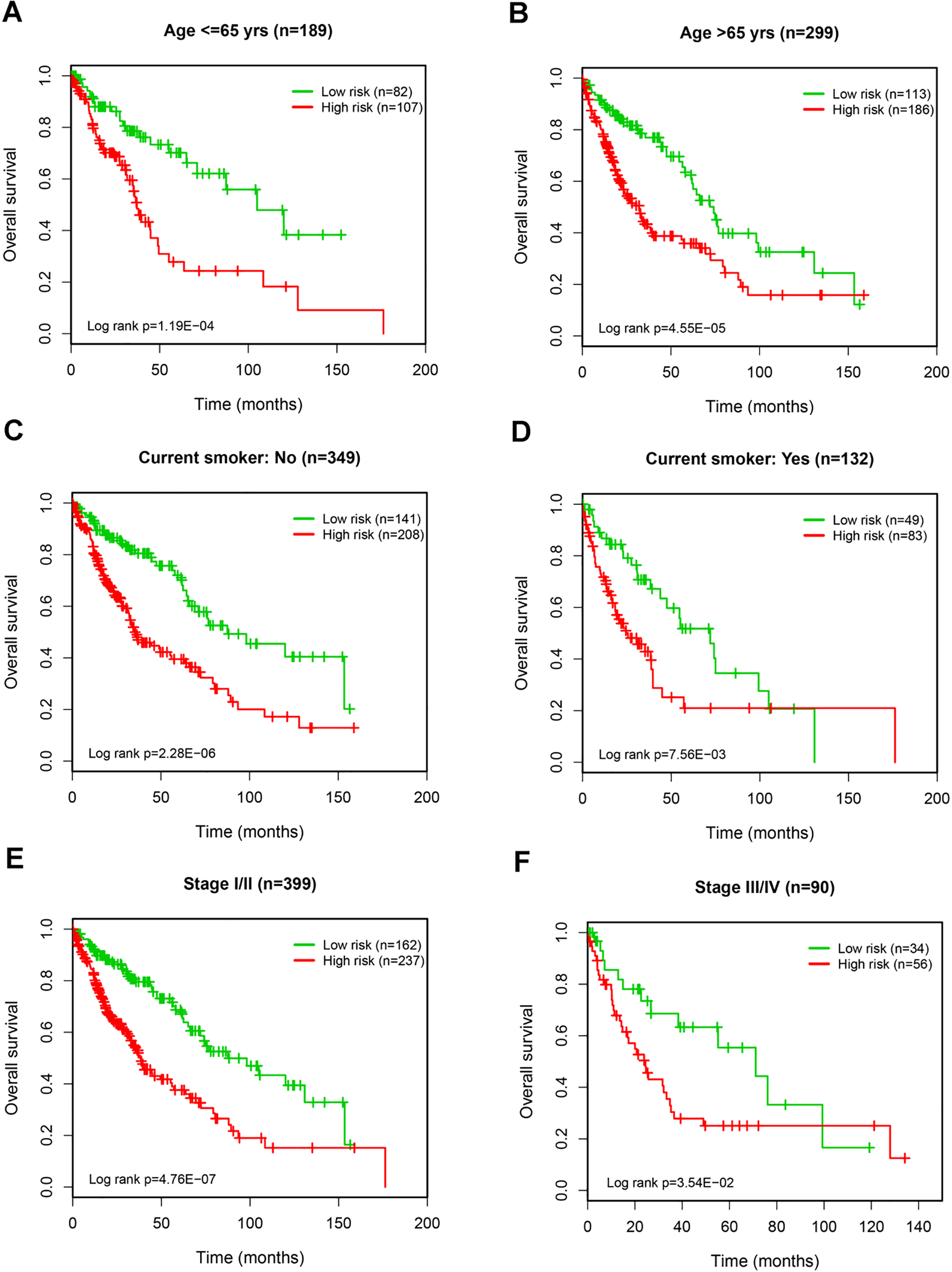
To assess the robustness of the 9-lncRNA signature in OS prediction of LUSC patients, we further examined it in the testing set and the entire set. The patients were divided into high- and low-risk groups using the same risk score model and cut-off point derived from the training set. The AUC of 5-year ROC curve in the testing and the whole sets was 0.613 and 0.705, respectively, implying a good prognostic capacity of the 9-lncRNA signature. Accordingly, patients in the low-risk group (
Independence of the lncRNA signature for survival prediction
Univariate and multivariate Cox regression analyses were conducted to test whether the prognostic ability of the 9-lncRNA signature was independent of clinical data. For each dataset, we included age, gender, smoking status, tumor stage, residual tumor and lncRNA risk score as explanatory variables. The results showed that the 9-lncRNA risk score was an independent predictor of OS (high- vs. low-risk group: HR
Potential biological roles of the lncRNA signature
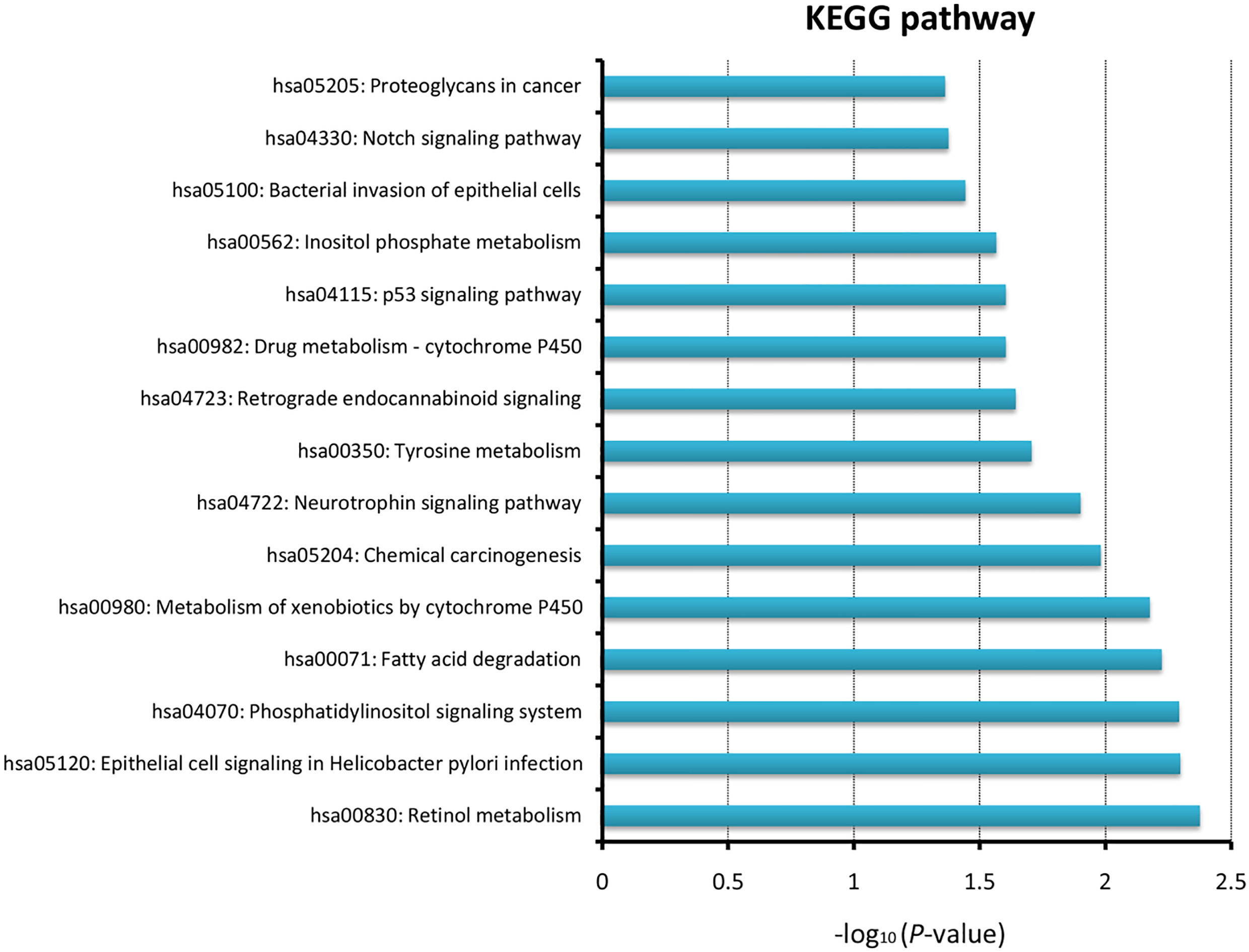
To investigate the potential functional roles involving the 9 prognostic lncRNAs, the co-expressed relationships between the lncRNAs and mRNAs were evaluated using Pearson correlation coefficients in the entire set. The expressions of 621 mRNAs were found to be highly correlated with at least one of the prognostic lncRNAs. KEGG pathways for these genes were then analyzed, using the whole human genome as the background. The results demonstrated that the prognostic lncRNAs were mainly enriched in pathways such as metabolism-related pathways, phosphatidylinositol signaling system, p53 signaling pathway, and notch signaling pathway (Supplementary Fig. 2).
Discussion
LUSC is a subtype of non-small cell lung cancer and accounts for nearly 40% of all lung cancer. There is a problem in not only the early detection but also the prognostic assessment of LUSC, which lead to a poor 5-year survival rate of the patients [1]. Although recent studies have improved the prognosis prediction for LUSC patients, most of them focused on biomarkers such as mRNAs or miRNAs. For example, Li et al. constructed a 4-mRNA signature to predict the outcomes of patients with LUSC [21]. Also, Gao et al. have identified a 7-miRNA signature that may have clinical implications in the outcome prediction of LUSC [22]. LncRNAs are a major subclass of ncRNAs expressed in a more tissue- and cancer-specific manner than mRNAs, implying their intrinsic utility in diagnosis and prognosis [23]. Indeed, several lncRNAs, such as VPS9D1-AS1 and MALAT-1, have been reported to be correlated with the survival of LUSC patients [24, 25]. However, to date, the prognostic lncRNA signatures for prognostic prediction of LUSC are scare and need to be systematically investigated.
In the present study, we analyzed the lncRNA expression profiles and clinical information of 493 LUSC patients from TCGA database. A set of 9 lncRNAs were identified to be strongly associated with patient’s OS by using univariate Cox regression and the subsequent RSFVH algorithm. These prognostic lncRNAs were then integrated into a 9-lncRNA risk score signature by combining their expressions and relative contributions in the multivariate Cox regression model. This signature showed a good performance in prognosis prediction, which could assign patients into low-risk and high-risk groups with significantly different OS in the training set. Similar predictive ability for OS of the 9-lncRNA signature was also documented in the testing set and the whole set. The above results suggested that the 9-lncRNA signature is reliable and robust in prognosis prediction of LUSC patients. In addition, by using the TCGA-LUSC dataset, a recent study has proposed a 6-lncRNA signature for outcome prediction of LUSC [26]. In that study, however, the 5-year AUC of ROC curve was only 0.672, indicating an inferior performance as compared to our 9-lncRNA signature.
To examine the predictive independence of the 9-lncRNA signature, we performed multivariate Cox regression analyses using the lncRNA risk score and available clinical data as explanatory variables. In the training, the testing and the entire sets, the 9-lncRNA signature was consistently identified to be an independent predictor for the OS of LUSC patients. Moreover, some clinical features, such as age, smoking status and tumor stage, were also significantly correlated with patient’s prognosis. Thus, to further confirm the utility of the 9-lncRNA signature in OS prediction, we carried out subanalyses by these clinical variables. We found that the prognostic power of the 9-lncRNA risk score was not modified by age, smoking status, and tumor stage. In summary, these findings indicated that the predictive value of the 9-lncRNA signature for OS of LUSC is independent of other clinical variables.
Although the number of documented lncRNAs has increased over the past decade, relatively few of them have been well-characterized. For example, only 184 human lncRNAs with known functions are included in the lncRNAdb v2.0 database [27]. It has been shown that lncRNAs exert biological functions through interacting with their partners such as mRNAs [28]. Therefore, the functions of the 9 prognostic lncRNAs were predicted by analyzing their co-expressed mRNAs. With functional enrichment analysis, we found that these lncRNAs mainly participate in regulation of metabolism-related pathways, phosphatidylinositol signaling system, p53 signaling pathway, and notch signaling pathway. Metabolism pathways, such as retinol metabolism, xenobiotic metabolism by cytochrome P450 and drug metabolism, have been reported to act as key enriched pathways in lung cancer patients [29, 30]. Recent studies have also highlighted the functional importance of phosphatidylinositol signaling system in LUSC [31, 32]. In addition to the well-known p53 signaling, alteration of the notch signaling pathway was increasingly reported as a pathological feature of lung cancer cells [33]. These results showed that the 9 prognostic lncRNAs may be involved in critical biological pathways associated with tumorigenesis and tumor progression of LUSC.
Some limitations should be acknowledged in this study. First of all, the 9-lncRNA signature was only identified and validated using RNA-Seq data from TCGA database. Therefore, this signature required additional confirmation in future large cohorts of LUSC patients. Second, although we used bioinformatics approach to explore the functional role of the 9 prognostic lncRNAs, the exact molecular mechanisms of them still remain unclear and need to be elucidated by experimental researches. Third, due to the lack of data, we cannot evaluate the impact of some confounders on the prognosis of LUSC patients, such as treatment strategies or medications.
Taken together, by analyzing the lncRNA expression profiles and clinical data of LUSC patients from TCGA database, we identified a total of 9 lncRNA biomarkers that were correlated with the OS of LUSC patients. The developed 9-lncRNA risk score model could effectively predict patient’s OS in the training set, which was further validated in the testing dataset and the entire set. Therefore, the 9-lncRNA signature may provide novel insights to predict the prognosis of LUSC patients.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary
A flowchart for data processing and analysis.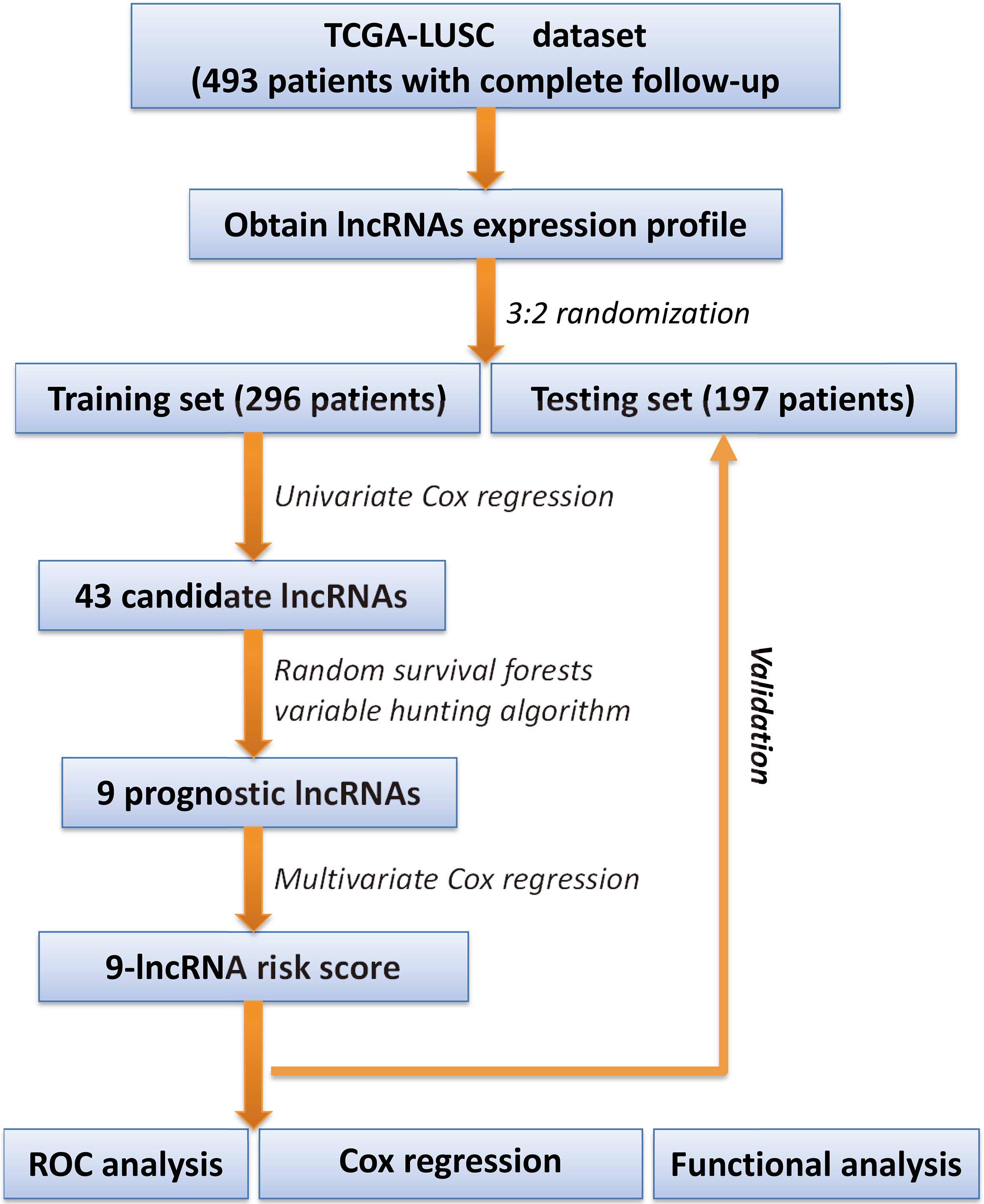
KEGG enriched by the 9-lncRNA signature using bioinformatics analysis.