
Abstract
BACKGROUND:
Prognostic biomarkers are promising targets for cancer prevention and treatment.
OBJECTIVE:
We try to filtrate survival-related genes for non-small cell lung cancer (NSCLC) via transcriptome analysis.
METHODS:
Transcriptome data and clinical information of Lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), mainly subtypes of NSCLC, were obtained from The Cancer Genome Atlas (TCGA) program. Differentially expressed genes (DEGs) analyzed by DESeq2 package were regarded as candidate genes. For survival analysis, univariate and multivariate Cox regression were applied to select biomarkers for overall survival (OS) and progression-free survival (PFS), where univariate analysis was for preliminary filtration and multivariate analysis considering age, gender, TNM parameters and clinical stage was for ultimate determination. Gene ontology (GO) analysis and pathway enrichment were used for biological annotation.
RESULTS:
We ultimately acquired a series of genes closely related to prognosis. For LUAD, we determined 314 OS-related genes and 275 PFS-related genes, while 54 OS-related genes and 78 PFS-related genes were chosen for LUSC. The final biological analysis indicated important function of proliferative signaling in LUAD but for LUSC, only cornification process had statistical meaning.
CONCLUSIONS:
We strictly determined prognostic genes of NSCLC, which would contribute to its carcinogenesis investigation and therapeutic methods improvement.
Introduction
Lung cancer has the leading morbidity and mortality among cancers, with about 2.1 million new cases and 1.8 million deaths estimated in 2018 worldwide [1]. The most frequent histopathological type of lung cancer is non-small cell lung cancer (NSCLC), comprising mainly lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) subtypes, on which mounting researchers lay great emphasis [2, 3].
With the comprehension of carcinogenesis advancing, therapeutic methods for NSCLC have achieved breakthroughs. The glamorous success must be assigned to targeted therapies based on tumor driver mutations, which could distinctly prolong survival periods in individual with specific genetic alterations compared to traditional platinum-based regimens [4]. Recently, immune checkpoint blockers have demonstrated powerful efficacy to patients carrying high expression levels of PD-L1(programmed cell death 1 ligand 1) or high tumor mutational burden [5]. However, there are still problems restraining from defeating such nightmare. On the one hand, a large portion of cases lack actionable mutations for targeted medicines, while many patients suffer from “cold tumors”, which are insensitive to current immunotherapies [6, 7]. On the other hand, drug tolerance comes to almost all patients sooner or later, inherent or adaptive resistance immensely weakens longtime benefits from targeted or immune therapeutic approaches [3, 8, 9]. So, molecular mechanisms underlying NSCLC still require deeper investigation.
Cancer-related gene remains as an important aspect for comprehending malignant etiology, while the first-line candidates are genes linked closely with clinicopathological characteristics of tumors. The Cancer Genome Atlas (TCGA) program contains sufficient tumors information including genetic and clinical records, which contribute enormously to investigation on cancers of many kinds [10, 11]. Here, by univariate and multivariate filtration, we finally obtained a series of tumor survival- related genes especially exhibiting independent risk suggestion, which will greatly enlighten pathogenesis investigation into NSCLC.
Materials and methods
Transcriptomic data and clinical records
LUSC and LUAD RNA-sequencing data and corresponding clinicopathologic annotation were derived from TCGA program [10, 11]. For LUAD, differentially expressed genes (DEGs) were found by comparing 58 tumors and 58 normal tissues, and 253 individuals containing transcriptome profiling and complete clinicopathologic information (age, gender, TNM parameters, clinical stage, overall survival status and progression-free survival status) were applied for survival analysis. As to LUSC, 51 tumors and 51 normal tissues were used to search DEGs and 288 individuals with analogous intact information were applied for survival analysis. Clinical characteristics of individuals contained in this research are shown in Table 1.
Clinical characteristics of individuals in TCGA-LUAD and TCGA-LUSC project
Clinical characteristics of individuals in TCGA-LUAD and TCGA-LUSC project
NSCLC-DEGs from TCGA RNA-sequencing data. (A) DEGs of LUAD; (B) DEGs of LUSC. DEGs, differentially expressed genes; NSCLC, non-small cell lung cancer; TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma.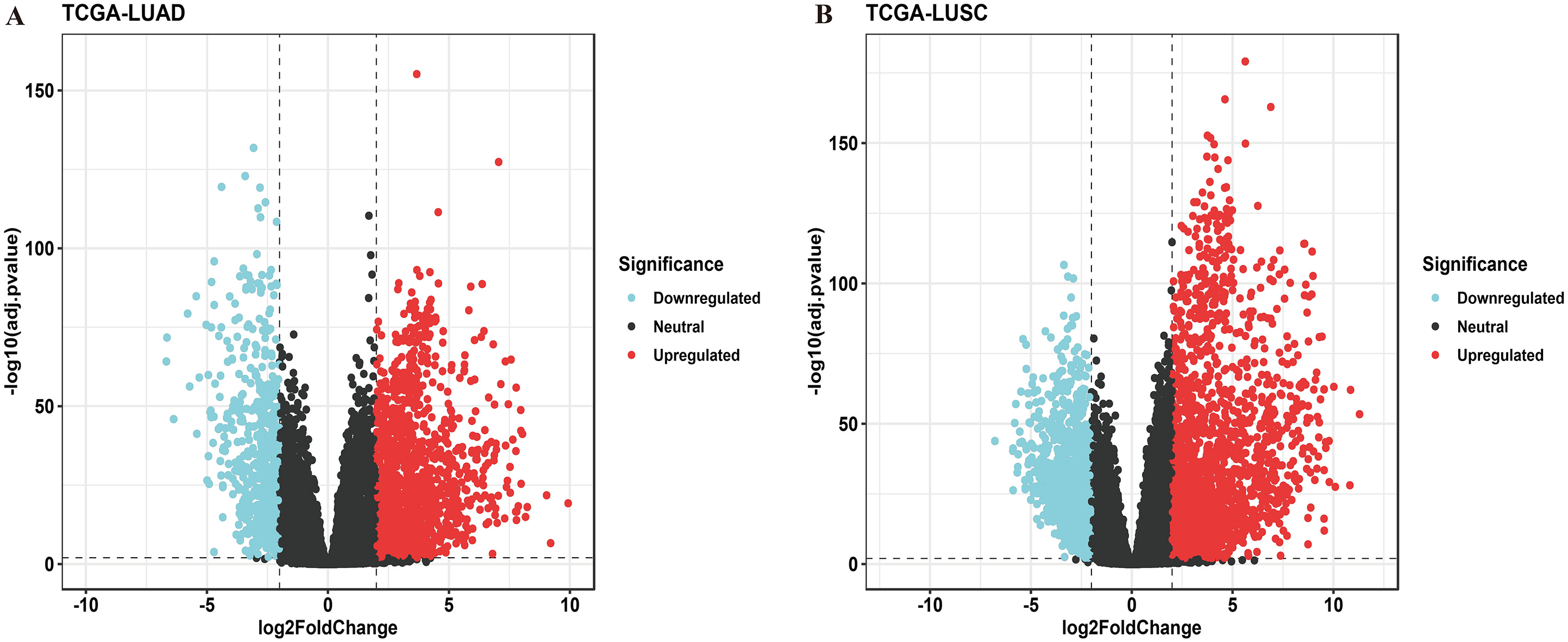
Biological signatures of NSCLC-DEGs. (A) gene ontology analysis of LUAD-DEGs; (B) pathway enrichment of LUAD-DEGs; (C) gene ontology analysis of LUSC-DEGs; (D) pathway enrichment of LUSC-DEGs. DEGs, differentially expressed genes; NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma.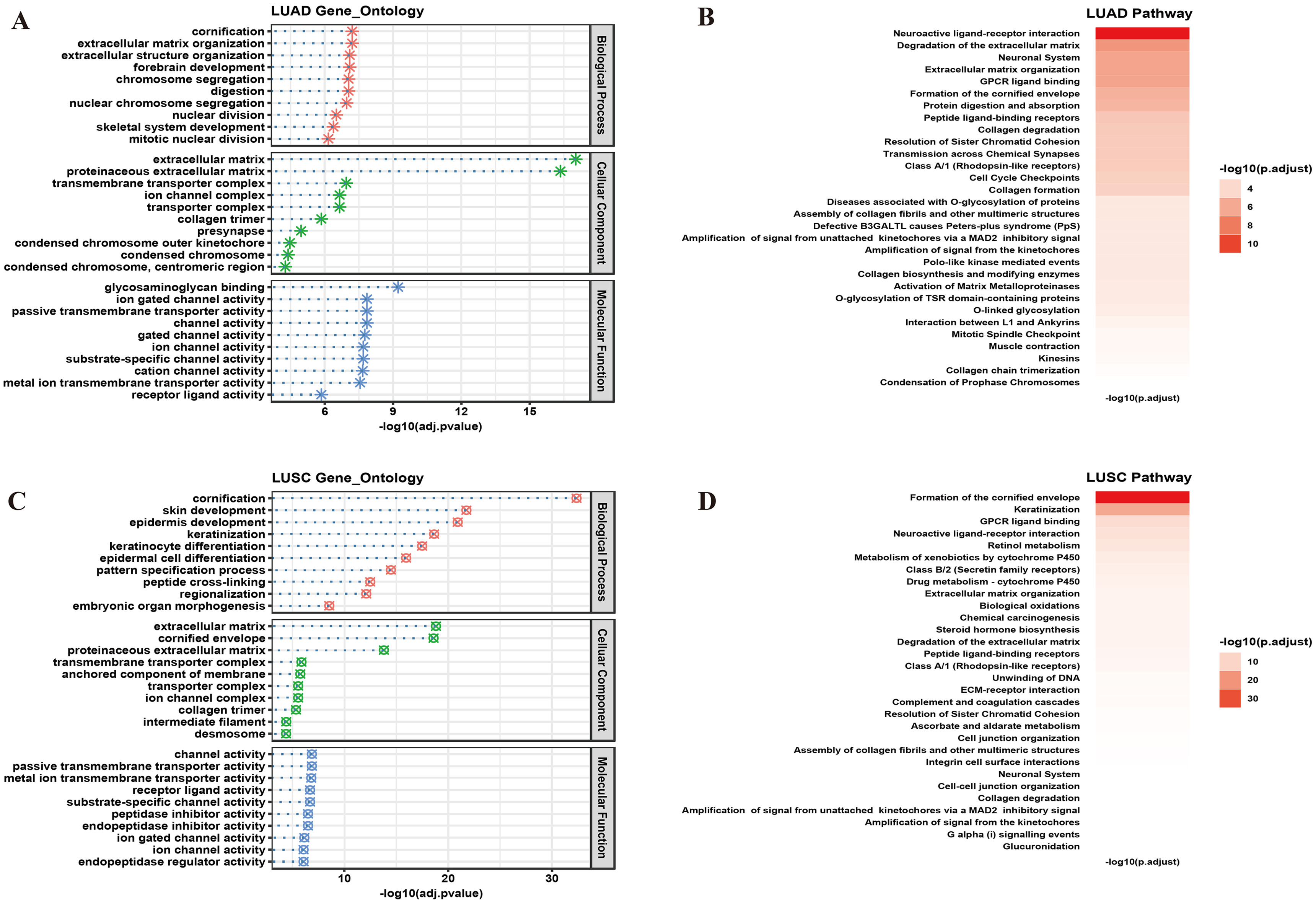
We first used BiomaRt and org.Hs.eg.db packages to map official genetic labels of these DEGs [12, 13]. Next we applied clusterProfiler package to execute gene ontology (GO) analysis involving molecular function, cellular component, and biological process [14]. Subsequently, we adopted clusterProfiler and ReactomePA packages to conduct pathway enrichment analysis based on Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome database [14, 15].
Selection of OS-related genes of NSCLC. (A) flow chart shows selecting OS-related genes of LUAD; (B) coefficients and 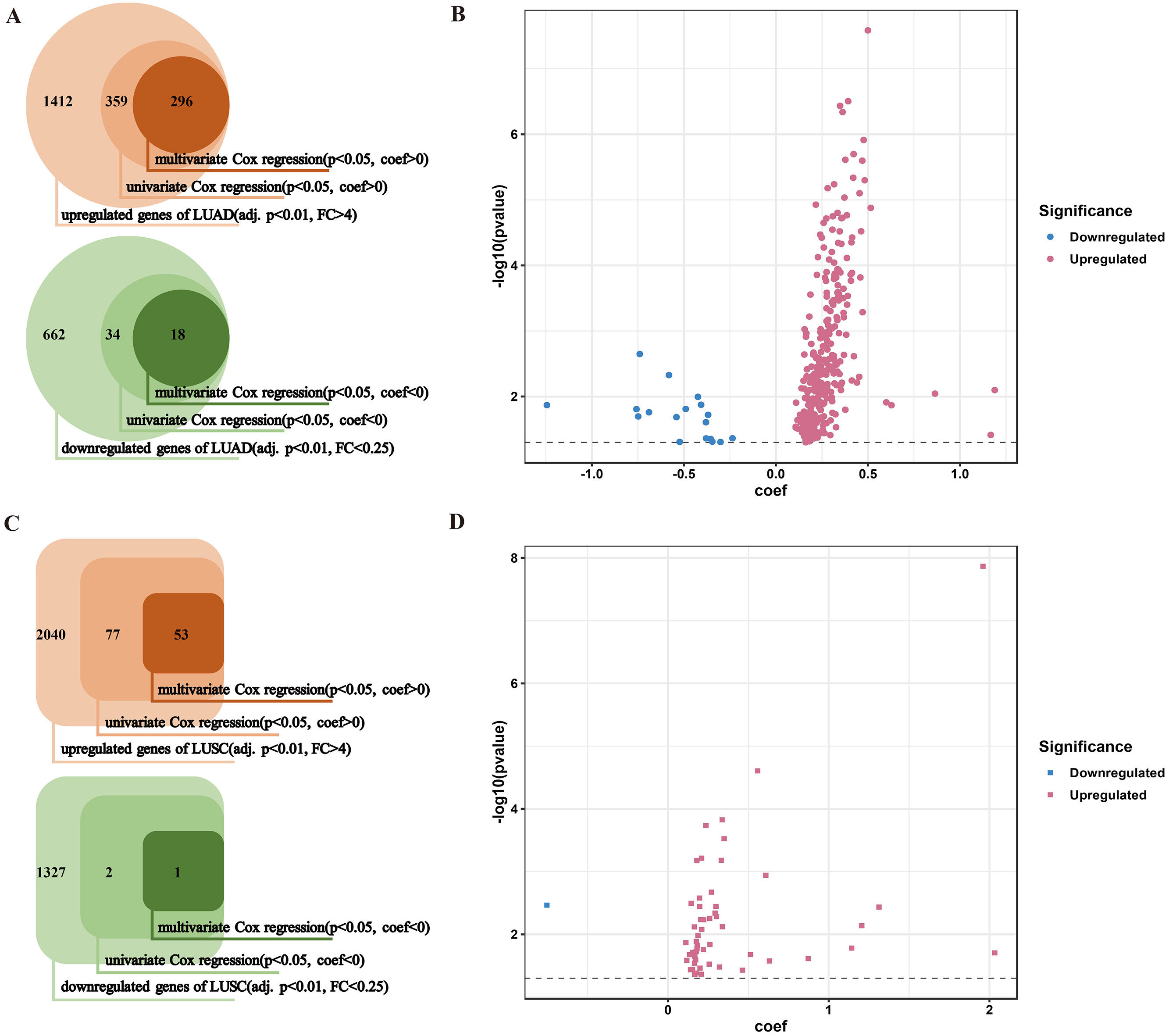
We used DESeq2 package to acquire DEGs from RNA-sequencing data (Adjusted
Results
Acquiring DEGs of LUAD and LUSC from RNA-sequencing data
Malignant progression largely relies on gene expression alterations, indicating momentous effect of DEGs upon carcinogenesis. Thus, we first tried to filtrate DEGs of LUAD and LUSC from public TCGA transcriptome data. DESeq2 package was practiced under strict inclusion criteria (fold-change
Filtration of PFS-related genes of NSCLC. (A) flow diagram shows selecting PFS-related genes of LUAD; (B) coefficients and 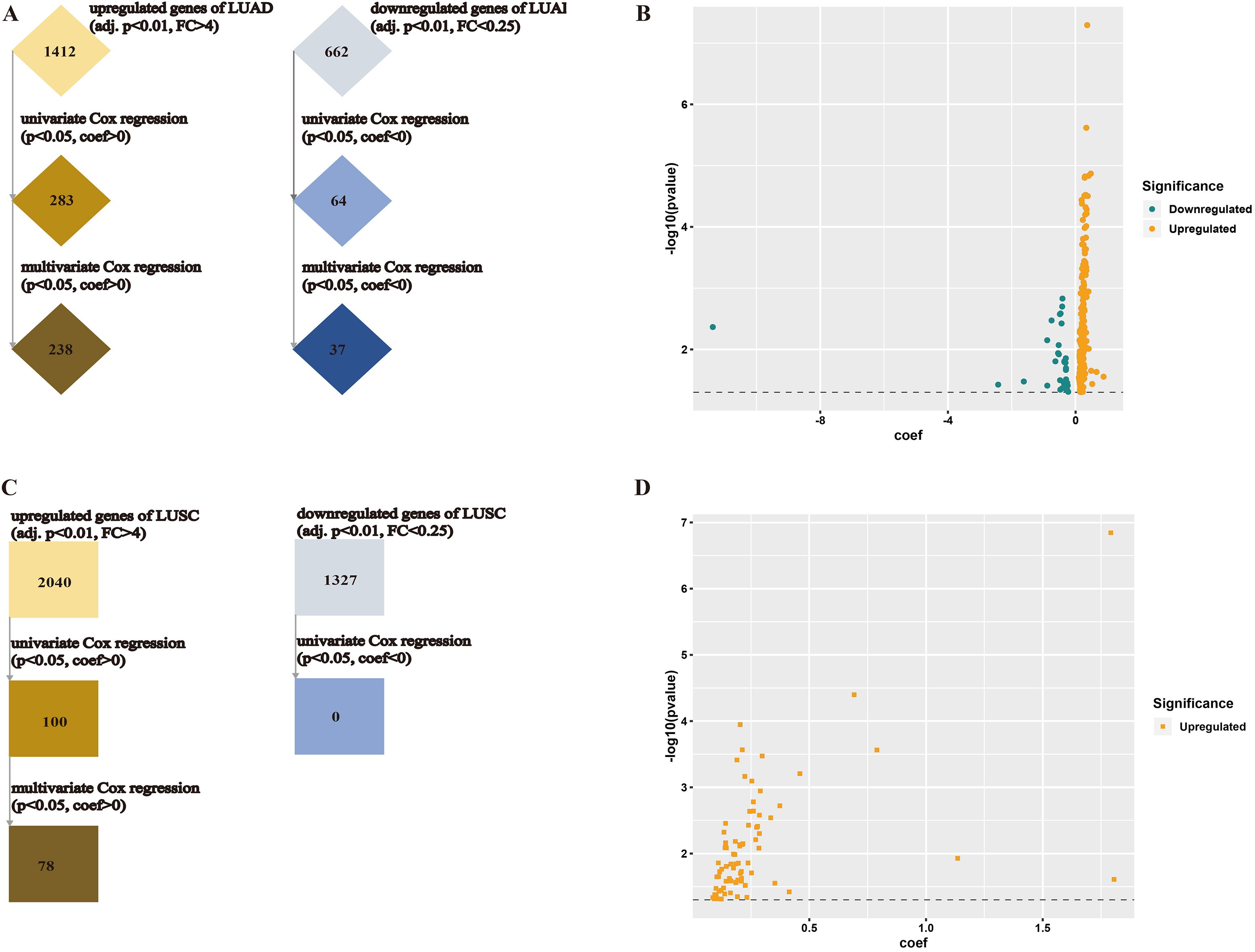
Biological annotation for prognostic genes of NSCLC. (A) Biological annotation for LUAD-related prognostic genes; (B) Biological annotation for LUSC-related prognostic genes. NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma.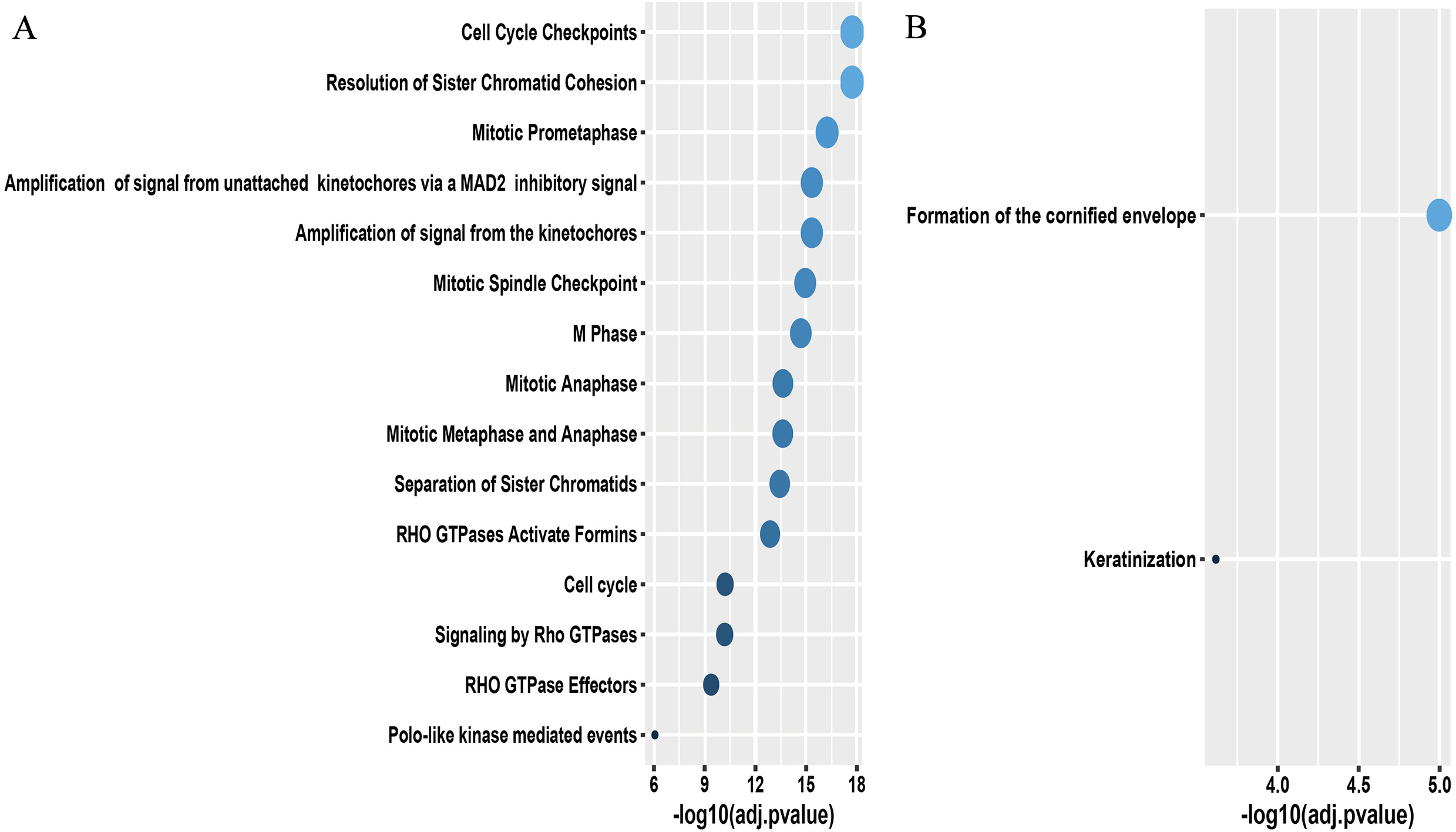
For further comprehension of these DEGs, we then investigated biological annotation via enrichment analysis. GO analysis and pathway enrichment analysis by clusterProfiler and ReactomePA packages were practiced to DEGs of LUAD and LUSC respectively. For LUAD, biological processes like extracellular matrix organization, collagen formation and degradation, cornification, ligand binding and ligand-receptor interaction topped the list (adjusted
Finding genes associated tightly with overall survival
Overall survival (OS) is an undoubtedly prominent indicators for cancer prognosis. We tried to dig out OS-related biomarkers by univariate and multivariate Cox regression based on statistic significance and regression coefficient (coef). For LUAD, 359 genes were first chosen from 1412 up-regulated DEGs (
Searching genes linked closely with progression-free survival
Progression-free survival (PFS) also imparts potent indications for cancer prognosis. We then applied the Cox proportional hazard regression to search biomarker of PFS, where univariate analysis was for primary filtration and multivariate analysis including factors like age, gender, TNM parameters and clinical stage was for ultimate selection. For LUAD, 283 up-regulated DEGs were chosen from univariate analysis and then 238 genes were filtrated by multivariate analysis (
Pathway enrichment analysis of prognostic DEGs
Subsequently, we investigated biological signatures of these prognostic DEGs by clusterProfiler and ReactomePA packages. And we found the prognostic DEGs of LUAD mostly converged on processes involving cell division and proliferation, while the prognostic DEGs of LUSC only had close association with cornification formation (Fig. 5A and B).
Discussion
Malignant progression is a dynamic biological process involving function of numerous genes. And rapid development in genomic and bioinformatic technology has provided much convenience to search cancer-related genes. In this research, we used bioinformatic tools to screen out genes closely linked to prognosis of NSCLC, providing much help for further investigation.
Cancer-related genes always exhibit aberrant expression in tumors compared to normal tissues. We first obtained DEGs from TCGA transcriptome data with critical standards set for high precision. Then we investigated biological characteristics of these DEGs, and we found different signatures of LUAD and LUSC. For LUAD, extracellular matrix (ECM) associated processes headed the list, suggesting that tumor microenvironment may contribute much to carcinogenesis of such histological type. And in fact, ECM alterations in the malignant stroma could function in kinds of aspects to facilitate neoplastic progression like promoting angiogenesis, assisting migration and invasion, providing sustainable signal for proliferation and survival, and so on [18, 19]. As to LUSC, most DEGs got enriched in biological sets of cornification and cell junction, which conforms to morphological characteristics of squamous cell carcinoma. Besides, cornification and keratins could not only been applied as diagnostic malignant markers especially about tissue origin, but also mediate diverse malignant properties such as metastasis, treatment responsiveness and the like [20, 21].
OS and PFS both are pivotal indictors for tumor prognosis, so DEGs linked closely with them always imply valuable information for handling cancer. Faced with quantities of DEGs, we conducted preliminary selection by univariate cox regression analysis, where admittance criterion was determined by
Cancer is a highly heterogeneous disorder and different histological type always exhibits some peculiar biological properties while possessing general malignant attributes [22, 23]. Noticeably, biological attributes of cancer-related genes we ultimately chose presented high consistence with known features of NSCLC. For LUAD, these genes were enriched in cell cycle and cell division activities. And as is well known, uncontrolled proliferation has always been a core hallmark of malignancy like LUAD, and many frequent driver mutations responsible for “oncogene addiction” like epidermal growth factor receptor (EGFR), Kirsten rat sarcoma (KRAS), tumor protein p53 (TP53) in LUAD are always involved in proliferative path-ways [24, 25, 26, 27]. As to LUSC, only the cornification process, a typical morphological feature of squamous cell carcinoma, but no signaling pathways proved to be significant. The result again revealed weakness in finding driving mechanisms of LUSC, and indicated molecular underpinnings facilitating LUSC might have big difference with canonical biological pathways contained in current public database [28, 29].
Certainly, our research has some drawbacks needed to be remedied in the future work. First, sample capacity should be enlarged to enhance precision for the high heterogeneity of NSCLC [23, 30, 31]. Second, posttranscriptional and posttranslational modification should be both considered for not just transcriptional regulation contributing to carcinogenesis [32, 33].
In conclusion, by screening out the whole transcriptome and considering typical confounding factors, we obtained a series of genes tightly associated with prognosis of NSCLC. And what we found would function as potential targets for etiology investigation and possibly be used as biomarkers for assisting clinical approaches of NSCLC.
Footnotes
Acknowledgments
We sincerely thanked Prof. Lei Shang and Prof. Lintao Jia for their professional guidance. This study was funded by the National Natural Science Foundation of China (81772462).
Conflict of interest
The authors have no conflicts of interest to declare.
Supplementary data
The supplementary files are available to download from