
Abstract
BACKGROUND:
Similarities between the pathologic progression of cancer and the physiologic process of placentation have been recognized for many years proposing that both present similar mechanisms and processes. Cervical cancer (CC) is one of the most frequent neoplasia among Mexican women turning it into an important health problem.
OBJECTIVE:
The aim of this study was to determine the degree of the involvement of pregnancy related genes and in cancer progression by in-silico analysis and validated in CC samples.
RESULTS:
The data mining analysis resulted in the identification of genes expressed in term placenta, first trimester placenta and normal cervical tissues. Finally, we selected KISS1 for the involvement of pregnancy related gene and also in cancer process. In order to explore KISS1 in CC, we analyzed Copy Number Variation (CNV) and gene expression using microarray experiments. KISS1 showed 20% genomic gain in 1q32.1 on CC samples. Furthermore, microarray analysis showed KISS1 as up-regulated genes. Results were validated showing an overexpression of 85% of KISS1 in CC samples.
CONCLUSIONS:
Data suggest KISS1 as a great candidate for CC molecular markers or as a therapeutic target for CC. Also, HPV presence does not seem to alter the KISS1 expression in CC.
Introduction
Cervical cancer (CC) has been established as a major gynecologic public health problem worldwide and the second most frequent neoplasm in Mexican women [1]. CC is the outcome of precursor lesions and presents risk factors such as smoking, hormonal contraceptives, multiple sexual partners, persistent Human Papilomavirus (HPV) infections and multiparity [2, 3].
Improvement in data mining such as, copy number variation arrays (CNV), expression microarrays and protein in public repositories which facilitate extraction of specific information using databases and on-line programs, have become important tools in the search for new molecular markers. The hallmarks of cancer have redirected research to finding mechanism such as inducing angiogenesis, activating invasion, evading immune system among other mechanisms [4] which are also present in pregnancy processes [5, 6, 7, 8].
For instance, placentation phenomena present in an analogous manner in malignant cells; placental trophoblasts migrates and invade the uterus to form new blood vessels. Another trait shared by cancer and the placenta is their ability to evade the immune response [6, 8] showing a significant similarity between a trophoblast and cancer cells [9]. In addition, there are reports that pregnancy hormones such as chorionic gonadotropin, estrogens, progesterone, also play a significant role in the development of some cancers such as breast, prostate, endometrial, and ovarian cancer [10]. In this context, genes present in both cancer and pregnancy processes can help identify new molecular markers and offer a different approach to cancer research. The objective of present study was to perform data mining of placenta and CC gene expression, looking for genes involved in both pregnancy and cancer. KISS1 participate during placental development, regulating trophoblast invasion [6] and participating in cancer [11, 12, 13, 14].
Work flow chart. Showing the protocol used for the search and download of a. SAGE libraries from CGAP and SAGEmap and b. CNV microarray datasets from 115 cervical cancer and 32 normal cervical tissues for gain and losses in chromosomes. c. Microarray datasets from GEO database (HG-U133 2.0 plus). *Cervical cancer expression and CNV microarray datasets as Marrero et al., 2017 described.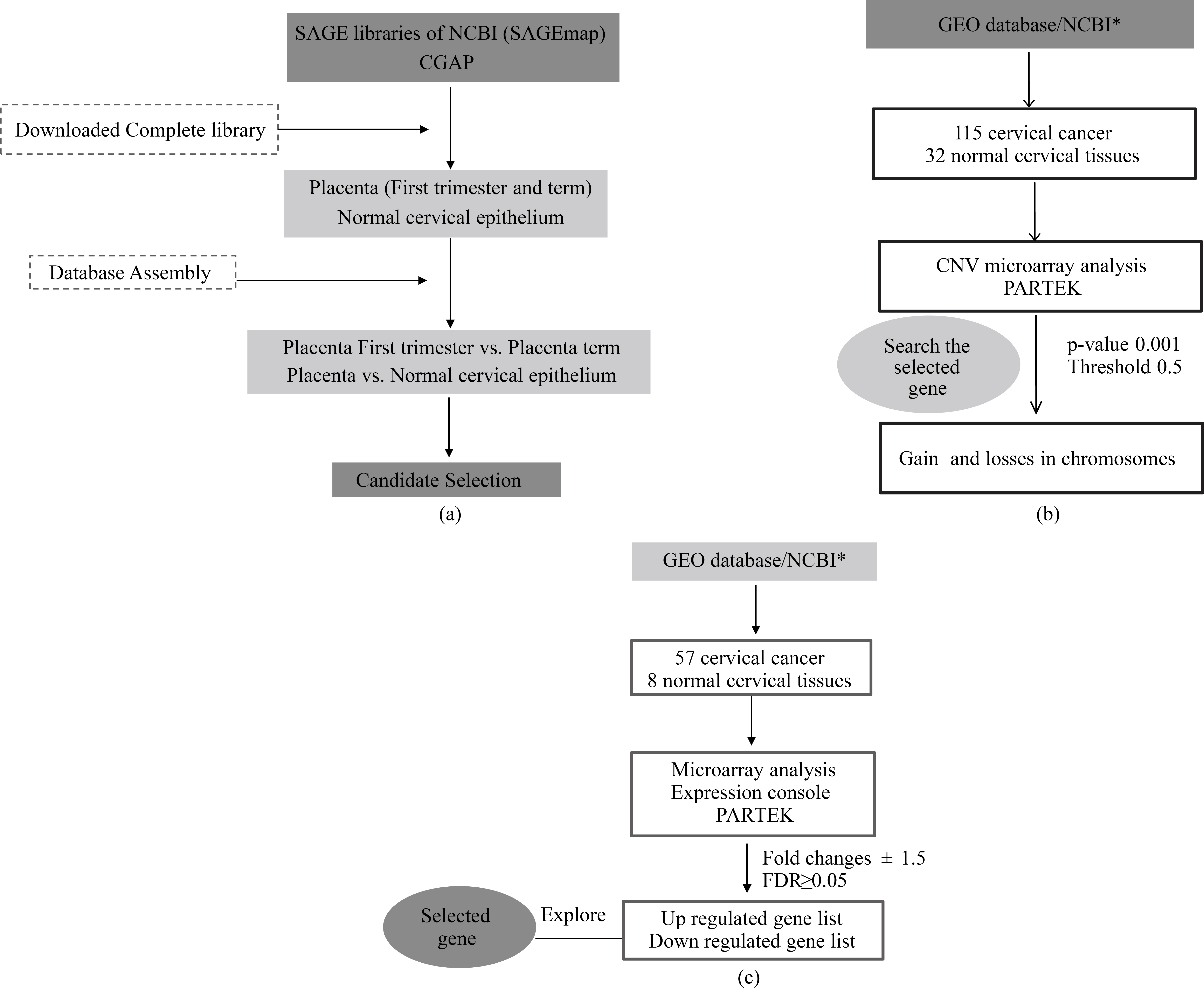
Gene selection criteria
The main selection criteria were genes present in placenta and CC samples, but not in normal cervical tissue. The data mining flux included in the present study is shown in Fig. 1.
Data mining with serial analysis of gene expression (SAGE) libraries
SAGE libraries were downloaded from Cancer Genome Anatomy Project (CGAP) and the National Center for Biotechnology Information’s SAGEmap (NCBI-SAGEmap). This study encompasses two libraries for placenta, first trimester (118,191 tags) and term libraries (89,414 tags), in addition to three different libraries for normal cervical epithelium data containing 30,418 [15], 165,624, and 181,224 tags respectively [16]. All the libraries were assembled and compared using Microsoft Office Access 2007. Candidate genes were selected under the criteria mentioned above.
Copy number variation data mining and analysis
Copy number variation (CNV) analysis comprised a total of 115 cervical cancer libraries and 32 control libraries corresponding to GSE10092 and GSE52904. Partek v6.6 (Partek Incorporated, Saint Louis, MO, USA) were used. The stringent parameters were set as follows: each segment must contain a minimum of 10 consecutive filtered probe sets, a p-value threshold of 0.001 when compared to the neighboring adjacent regions, and a signal-to-noise threshold of 0.5. The cut-off value for the gain was set at above 2.3, while loss was set at below 1.7. CNV was called for the gains or losses that occurred in at least 10% of the total samples. The 22 somatic chromosome and sexual X were analyzed. Hierarchical clustering was performed by the following parameters, sample dissimilarity: Euclidian and cluster method: average linking (Marrero-Rodriguez et al., 2017, submitted).
Cervical cancer transcriptome in-silico analysis
The selected genes from SAGE analysis were also subjected to microarrays analyses. A total of 8 normal (N) and 57 cervical cancer (CC) experiments were downloaded from European Bioinformatics Institute (EMBL-EBL) and GEO. These data GSE7307, GSE5787, GSE3526 and Gene Expression Atlas(GSE2109) corresponding to Affymetrix Human Gene Chip U133 plus 2.0 platform.
Data sets analyses were achieved by means of CEL files with the Partek Genomics Suite 6.6v software (Partek Incorporated, Saint Louis, MO, USA), Expression Console and Transcriptome Analysis Console (Affymetrix, Santa Clara, CA, USA). Pearson and Spearman correlation was performed and probe sets were summarized by means of Median Polish and normalized by quantiles with no probe sets excluded from analysis. Background noise correction was achieved by means of Robust Multi-chip Average (RMA) and data were log2-transformed. Data grouping and categorization was achieved by principal component analysis (PCA). Differentially expressed genes were detected by means of ANOVA. Genes were considered altered with
Cell culture conditions
The following human cervical cancer cell lines were studied and analyzed: CaSki and SiHa (HPV16); HeLa (HPV18); C33A (HPV-negative). These cell lines were growth in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Life Technologies, CA USA) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% Penicillin-Streptomycin (Gibco) at 37
Sample collection
The clinical samples used in the present work were previously used [17]. Twenty cervical cancer samples were collected from the Hospital General at Clinica de Displasias in Mexico City. All participants signed for informed consent and ethical approval was granted from the institutional board in accordance to the declaration of Helsinki. Detailed clinicopathological information was obtained from patients records. All samples were collected in RNA later solution (Ambion, CA, USA) and prior to the administration of any chemotherapy and/or radiotherapy treatments. Eighteen normal cervical tissues were collected from patients who had undergone some type of hysterectomy procedure with healthy cervix who accepted to participate were used.
RNA extraction
Tissue homogenization was done in TissueLyser (QIAGEN, Germany) with TRisure reagent (Bioline, UK). Once homogenized, chloroform was added and samples were centrifuged at 8,000
DNA Extraction and HPV genotyping
The organic phase from RNA extraction was used for DNA extraction. Absolute ethanol was added to the organic phase and centrifuged 5 min at 8,000
cDNA synthesis and end-point PCR
cDNA synthesis process used Superscript Vilo Master Mix (SVMM) (Life Technologies, USA) were used according to manufacturer’s condition (RNA 1
RT-qPCR
For RT-qPCR, TaqMan Gene Expression probe for KISS1: Hs00158486_m1 (Applied Biosystems, MA, USA) and RPLP0: Hs_99999902_m1 (as housekeeping gene) were used. All PCRs were performed using Applied Biosystem StepOne™Real-Time PCR System with conditions specified by the manufacturer, also all reagents were purchased from Applied Biosystems (MA, USA), and subjected to the following conditions: 11
Immunohistochemistry assay
A tissue microarray including 38 cases (20 CC tissues and 18 normal cervix) was constructed from paraffin-embedded sections, formalin-fixed tissueblocks were stained with haematoxylin and eosin therefore reviewed by a pathologist to select areas of invasive tumor. Core samples were taken using 0.6 mm
Ten most frequent tags in first trimester and term placenta
Ten most frequent tags in first trimester and term placenta
*KISS1 gen tag as one of the highest frequency per million in first trimester and term placenta.
The cell lines (SiHa, HeLa and C33A) were grown on glass cover slips pre-treated with poly-D-Lys. The cells at 70% of confluence were fixed in 4% paraformaldehyde (in PBS 1x pH7.4 for cell lines). The fixed cells were washed with PBS 1x (3 times). Cells were permeabilized with PBS 1x-Triton 0.2% (200
Statistical analysis
Clinical and pathological relation analysis was performed by means of the Student’s t test for KISS1 expression. All p values represent two-tailed tests and were considered significant at 0.05. Statistical analysis was performed using SPSS v15 statistical software.
Results
SAGE libraries for placenta and CC
SAGE data were mined for highly expressed genes in placenta but not in normal cervical tissue. Only genes with 10 or more tags per million were selected resulting in primary selection of 1587 genes expressed in term placenta, 1057 genes in first trimester placenta, and 345 genes in normal cervical tissues From these, only 50 highly expressed genes from the placenta were selected. Finally, the selection was narrowed down to only 10 genes that were present in placenta but not in normal cervical SAGE libraries were selected. During the selection process, KISS1 gene (Table 1 and Fig. 2) was identified as one of the most frequent tags in placenta but not in normal cervix.
1q32.1 gain in CC, KISS1 copy number variation
Our first approach to analyzing KISS1 copy number variation was to identify cytogenetic regions harboring DNA gains. Results revealed that there were representative regions such as 1q, especially 1q32.1 where IL20 and MDM4 genes are encoded, with over representation in
Data mining for placenta and cervical cancer sample. A. Comparison of SAGE libraries; placenta first trimester/term vs. normal cervix for KISS1. KISS1 tag was absence in normal cervical epithelium. B. Hierarchical cluster for Chromosome 1, gain and losses in 115 cervical cancer samples.
C. Heat map of 65 samples of CC and normal cervical samples.
KISS1 expression in cervical cancer cells. a. Expression of KISS1 in cervical cells lines. b. Expression of KISS1 gene in 20 samples of CC (1–20), two healthy cervix samples (N1 and N2) and RPS18 were used as housekeeping gene. KISS1 expression in all CC sample were observed with different intensity of band. c. Relative expression of KISS1 in CC and normal cervix samples with RT-qPCR with heat map of KISS1 expression for each sample. KISS1 overexpressions in CC were 80% and in normal tissue absence of KISS1 were observed. Also, HPV infection were present in 85% of CC sample and 65% were simple infection and 20% were multiple infection.
Immunodetection of KISS1 in Cervical Cancer cells. A. Immunofluorescence assay for Kisspeptin in HeLa, SiHa and C33A, DAPI counterstained x1000 original amplification. B. Immunohistochemical staining of KISS1 in normal cervical tissue (1) and tumor tissue; squamous carcinoma (2) adenocarcinoma (3) and term placenta (4), Haematoxylin counterstained x40 original amplification. Immunodetection of KISS1 was independent of histological classification and HPV genotype.
To explore KISS1 gene expression in CC, gene expression microarrays from CC data were used. Lists of up and downregulated genes were generated (Fig. 2). Interestingly, the KISS1 gene was one of the up-regulated genes found among 11,338 genes. To validate the present bioinformatics data mining we proceeded to analyze KISS1 expression in cervical cells.
KISS1 gene expression in CC cells
Total RNA from cervical cancer cell lines were subjected to RT-PCR analysis as a CC study model. We observed that KISS1 gene expressed in all four cell lines used. Normal cervical sample showed negative KISS1 expression and CC samples presented KISS1 gene expression in different band intensity (Fig. 3). Encouraged by these results, real-time RT-PCR was performed to determine the relative expression of KISS1 in a subset of 20 samples: 4 adenocarcinomas and 16 squamous carcinomas. Three samples of normal cervical tissue were used to RT-qPCR and, as expected; an absence of KISS1 gene expression was confirmed while in CC samples 80% showed de novo expression with more than 10-fold change (Fig. 3).
HPV Genotyping
Eighty-five percent of CC samples were HPV positive, and genotyped detecting HPV 16 in 35%, HPV 18 in 10% HPV 31, HPV 51, and HPV 66 in 5% each, non-identified HPV in 5%, and three multiple infection in 20%, discarding HPV infection might not influence KISS1 gene expression.
KISS1 Immunodetection
Immunoflourescence detection on three cervicalcancer cell lines was evaluated to discard any influence of HPV in KISS1 expression. All three cell lines showed positive cytoplasmic immunodetection of KISS1. Immunohistochemical analysis of patient tissues involved the evaluation of immunostaining for negative and positive staining. All normal cervical samples presented negative expression in cytoplasm and nuclei, unlike CC samples which presented strong cytoplasmic immunostaining in epithelial cells without exception (Fig. 4). Positive immunodetection occurred regardless of the presence of an HPV infection or its genotype.
Clinicopathological correlation with KISS1 expression
KISS1 expression in CC sample was statistically significant with
Discussion
The correlation between KISS1 expression and clinicopathological parameters
All the
In the present work, data mining was performed to identify expressed genes related to cancer and pregnancy in cervical and placental tissues. KISS1 was expressed in placental and cervical cancer tissues but absent in normal cervix tissue. The selected KISS1 gene, which is located on chromosome region 1q32.1, has been studied in several types of cancer and has been suggested to play several roles in cancer [19]. For example, the absence of KISS1 gene expression has been associated with increased metastasis and cancer progression in bladder [20], ovarian [21] and pancreatic cancer [12]. In contrast, an increased KISS1 expression is related to metastatic capacity in breast [22] and hepatocellular carcinomas [23]. This suggests that KISS1 gene could be a dualfaced gene in cancer and its tissue specific function. Also, KISS1 gene is highly expressed in the placenta, nervous system, testis, ovary, pancreas and intestine. KISS1 participation in pregnancy is associated with the process of placentation and trophoblast invasion [8]. KISS1 protein regulates the invasion of trophoblast into maternal deciduas, essential for fetal development [6] and presents similarities with the invasion process of cancer cells [5]. This could support the biological point of view that trophoblast invasion sharing common features with tumor invasion exploiting similar molecular mechanism [9]. A gain of 1q32 was observed in 20% of CC sample as its reported [24]. Therefore it could suggest that there is no relationship between gain of 1q32.1 and KISS1 expression and that might be regulated by an epigenetic mechanism. There are some reports of KISS1 regulation through a methylation mechanism [14, 25], although further experimentation is required to corroborate this mechanism in CC Cervical cancer cell lines were used as a model for our first approach, observing KISS1 expression in the cell lines with or without HPV sequences and suggesting that KISS1 expression could be an event independent from HPV infection or genotype. Similar results were also observed on CC patient samples regardless of the histology type and HPV status. Positive cytoplasmic microvesicles KISS1 expression was observed during research, suggesting that KISS1 may participate in calcium mobilization within CC as described in different tissues [19]. This calcium mobilization could stimulate extracellular signal-regulated kinase-1 and -2 (ERK1/2) and mitogen-activated proteins (MAPKs) [26].
We observed KISS1 expression in CC sample10 times more in comparison to normal cervical tissue, having de novo overexpression of the KISS1 gene. Particularly, one sample from patient with uterine myomatosis (data not shown), which was discarded, displayed low KISS1 expression suggesting hormonal misbalance interference in via estrogen receptor (ER) as described [27]. We are also considering increasing the number of normal samples, to support this preliminary result.
There is one small-molecule, Honokiol that has been used as an anticancer agent and it evidence supports the notion that it suppresses KISS1/KISS1R signaling in renal cell carcinoma [28]. It would be interesting to study the Honokiol effects in CC cells and also it might propose as a potential CC therapy.
Finally, the present work could represent an innovative approach searching cancer markers (embryonic antigens). The expression of KISS1 in CC suggests that this gene may be great candidate for molecular marker and/or therapeutic target for CC. Furthermore additional studies are required to elucidate the KISS1 role in CC and it is been analyzed the precursor lesion for CC (in preparation); both matters are currently under investigation by our work team of researchers.
Footnotes
Acknowledgments
This work represents partial fulfillment of the requirements for the Ph D. degree for KTP at Doctorado en Biomedicina y Biotecnología Molecular, ENCB-IPN. During this work, the authors, KTP, DMR, MMR, VHP, NM, GPN, HA and MRE were recipients of a Consejo Nacional de Ciencia y Tecnologia (CONACYT) fellowship and grant 202222 from Fondos Sectoriales. MS was a Fundación IMSS fellowship recipient. RMR-A is recipient of COFAA-IPN and EDD-IPN fellowships.
Conflict of interest
All other authors declared no conflict of interest.