
Abstract
Cervical cancer is one of the leading causes of death in women worldwide, which mainly affects developing countries. The patients who suffer a recurrence and/or progression disease have a higher risk of developing distal metastases. Proteases comprising the degradome given its ability to promote cell growth, migration, and invasion of tissues play an important role during tumor development and progression. In this study, we used high-density microarrays and quantitative reverse transcriptase polymerase chain reaction to evaluate the degradome profile and their inhibitors in 112 samples of patients diagnosed with locally advanced cervical cancer. Clinical follow-up was done during a period of 3 years. Using a correlation analysis between the response to treatment and the development of metastasis, we established a molecular signature comprising eight degradome-related genes (FAM111B, FAM111A, CFB, PSMB8, PSMB9, CASP7, PRSS16, and CD74) with the ability to discriminate patients at risk of distal metastases. In conclusion, present results show that molecular signature obtained from degradome genes can predict the possibility of metastasis in patients with locally advanced cervical cancer.
Keywords
Introduction
Cervical cancer (CC) is the second most common cancer in women worldwide with 275,000 deaths estimated in 2008. Developing countries concentrate on almost 88% of the diagnosed women. CC is detected by simple cytological screening methods such as “Pap smear,” which continue to be successfully employed in advanced countries. However, in less developed regions, most patients are diagnosed in locally advanced stages associated with the presence of large tumors whose mortality/incidence ratio is about 50%.1,2 The standard or conventional treatment for patients with locally advanced cervical cancer (LACC) classified as IB2-IVA is based on radiotherapy in combination with cisplatin chemotherapy (40 mg/m2), followed by brachytherapy.3,4 Despite the standard treatment, approximately 40% of patients develop distal recurrence that is a clinical manifestation of a metastatic disease (MD).
The conventional treatment in LACC offers clinical benefits by controlling pelvic tumor growth. However, different studies revealed that the treatment does not extended the overall survival in at least 40% of patients,5,6 and also, that more than 35% of patients can develop a distal recurrence. 7 The most common organs affected by distal metastatic growth are liver, lungs, paraaortic lymph nodes, and bones. 8 Therefore, it is imperative to discover and validate molecular biomarkers which may be used for early detection of distal recurrence, hence allowing physicians to provide individual alternative treatments as soon as possible. The arrival of high-throughput technologies enables us to define tumors as a function of gene expression profiles and use this information to improve identification of patients who would benefit with the conventional treatment and those who will develop distal recurrence.
Despite clinical significance of metastasis in cancer patients, the molecular processes involved in migration and invasion of cancer cells are poor understood. However, it is clear that a central mechanism is activated by metastatic cancer cells, producing degradation of extracellular matrix (ECM).9,10 Many research groups have focused on showing the participation of multiple ECM proteases and their inhibitors as regulators and mediators of metastatic behavior in CC tumors and derived cell lines.11,12 We reasoned that degradome-associated genes have an important role in cancer metastasis and cellular migration process in LACC patients.
The human degradome encompasses the entire genes that encode proteases (also recognized as peptidases and proteases) and their endogenous inhibitors, known as proteases inhibitors.13–15 The biological and physiological meaning of the degradome is evident in several key processes such as embryonic development, cellular differentiation, proliferation, apoptosis, migration, angiogenesis, and malignant invasion. 13 At present, several studies have analyzed the expression of specific proteases and their inhibitors in the context of clinical information in CC patients, and particularly it has been shown in different studies that matrix metalloproteinase (MMP)-2 and MMP-9 are overexpressed in CC specimens, in contrast to normal counterparts.11,12 However, the degradome-related genes have not been analyzed yet by a global and systematic approach in CC and their association with clinical response.
In this report, we aimed to evaluate expression levels of the entire protease family consisting of cysteine, aspartyl, threonine, metallo and serine proteases and their inhibitors in a cohort of 112 patients diagnosed as LACC using genome-wide high-density microarrays. We found that 562 genes were aberrantly expressed in tumor tissues (373 genes were overexpressed and 189 downregulated in tumors with respect to normal tissues). After conventional treatment, patients were followed up by computerized axial tomography scan (CAT scan) and then assigned to complete responders (CR) or MD patients who developed new target lesions on distal organs according to RECIST (Response Evaluation Criteria In Solid Tumors) 1.1 criteria. Our findings revealed eight degradome-related genes with the ability to predict distal metastasis in patients with LACC.
Materials and methods
Patients
This study has been approved by the research and ethics committees of the National Cancer Institute of Mexico (INCan) and has been conducted in agreement with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments. A total of 112 patients diagnosed as LACC were prospectively registered in the INCan tumor-banking protocol at the time of diagnosis (April 2010 through August 2012). Immediately after punch biopsy, tumor samples were split into three pieces, one for pathological confirmation of at least 80% of tumor cells and the remaining two for RNA and DNA isolation, respectively. RNA and DNA biopsies were frozen in liquid nitrogen until nucleic acid extraction. The patient eligibility criteria consisted as follows: (a) confirmed pathologic diagnosis of CC staged IIB and IIIB (LACC), (b) biopsies with pathology report with more than 80% of tumors cells, (c) age range 20–70 years, (d) high-quality DNA and RNA samples, (e) no other comorbidity, (f) no previous oncological treatment, and (g) patients able to receive the standard or conventional therapy based on concurrent chemotherapy and radiotherapy. Chemotherapy was based on weekly CDDP (cis-diamminedichloroplatinum (II)) at 40 mg/m2 during five–six cycles. Radiotherapy consisted of external radiation and intracavitary brachytherapy, for a total dose of 64–66 Gy over 67 days. 4 All patients received the same conventional treatment. Clinical characteristics of patients are summarized in Table 1. Healthy cervical tissues were obtained from patients who had undergone hysterectomy by uterine myomatosis. Inclusion criteria are as follows: (a) no previous cervical surgery (such as the loop electrosurgical excision procedure or cone biopsy), (b) no human papillomavirus (HPV) infection, (c) no hormonal treatment, and (d) at least three previous negative Pap smears.
Clinical characteristics of all patients.
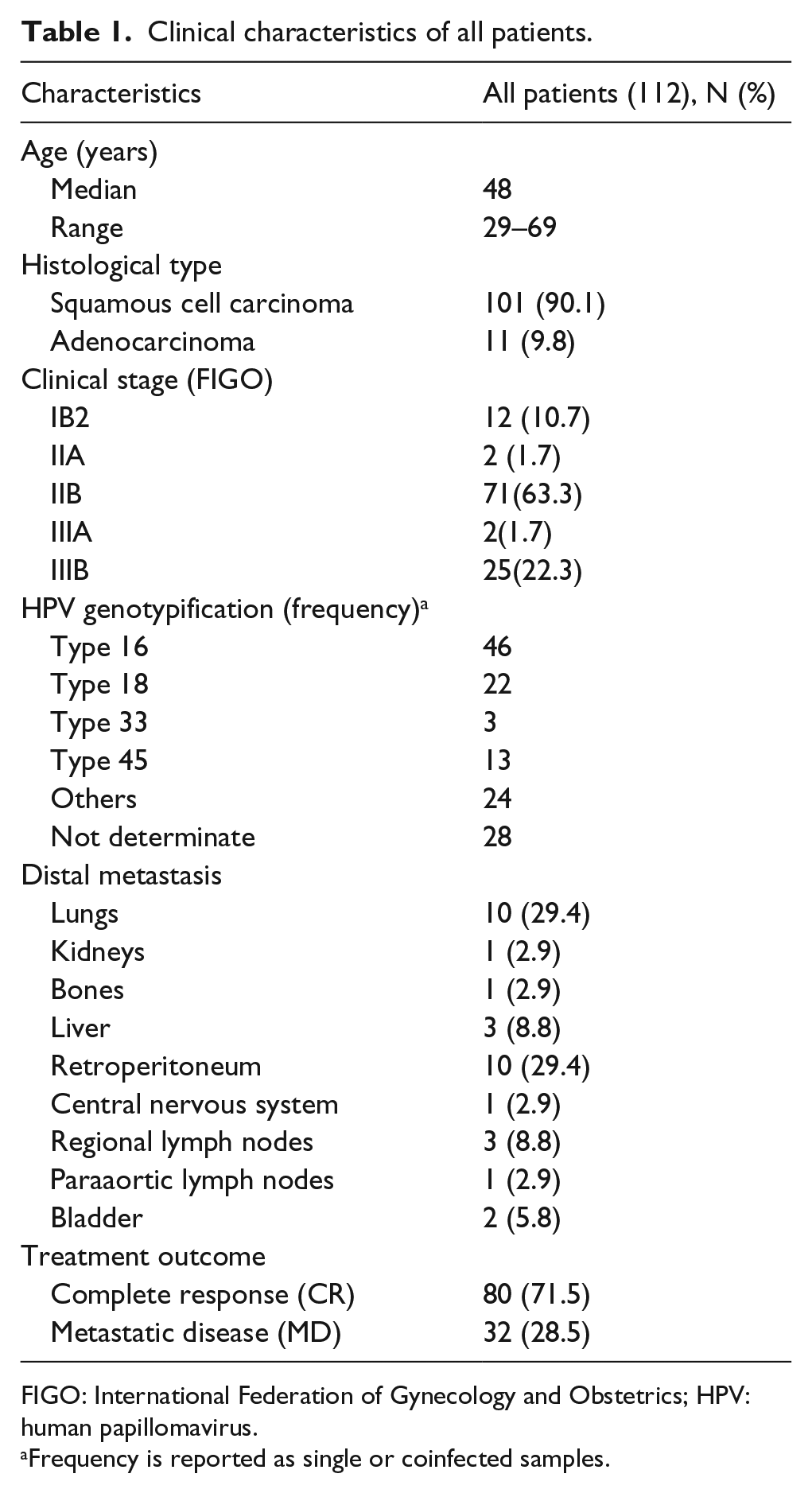
FIGO: International Federation of Gynecology and Obstetrics; HPV: human papillomavirus.
Frequency is reported as single or coinfected samples.
Clinical definitions
The staging was assessed according to the International Federation of Gynecology and Obstetrics (FIGO) classification. Clinical response was evaluated by RECIST 1.1 criteria and CAT scan. CR was recognized as the disappearance of all signs of cancer in response to treatment. The appearance of one or more new lesions after treatment was considered as MD. 16
HPV genotyping
DNA was obtained from cervical tumor biopsies using MagNAPure Compact Instrument following the manufacturer’s recommendations (Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany). HPV genotyping was assessed by two approaches, Linear Array HPV Genotyping (Roche Diagnostics GmbH, Roche Molecular Biochemicals) and Nested Multiplex Polymerase Chain Reaction (NMPCR MY/GP primers) with subsequent PCR-fragment direct sequencing. 17
RNA purification and microarray hybridization
A total of 85 samples were obtained at the time of diagnoses and were used to discover a gene degradome-related expression profile. We compared gene expression signatures of patients with CR and gene expression signatures of patients diagnosed with MD. The quality of RNA was evaluated through 18S:28S ratio. Hybridization targets were prepared from 250 ng of total RNA and amplified with Whole Transcriptome Amplification Kit 2 (Sigma-Aldrich, St Louis, MO). Four micrograms of amplified and Cy3 labeled complementary DNA (cDNA) were used to hybridize into high-density microarrays containing 45,000 features according to the recommended protocol of Nimblegen (Nimblegen Roche, Mannheim, Germany). After standard washes, the arrays were scanned on the Nimblegen MS200 microarray scanner. Digitalized images were stored for further analyses.
The raw microarray data of this study is publicly available at GEO database (Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE56303. 18
Microarray preprocessing and statistical analysis
Digitalized images were gridded by using the NimbleScan v2.6 Software (Nimblegen Roche). Then, robust multi-array analysis background normalization and quantile normalization were performed for intra- and inter-array normalization, respectively. Genes with signal intensities above 95% of arbitrary threshold were filtered. 19 Differential expression between normal and tumor tissues and between clinical outcomes was assessed by analysis of variance (ANOVA) tests and significance statistics for each gene were obtained by Genesis software (ver. 1.7.7). 20 Using the same software, graphics were generated.
Expression of degradome-related genes
To evaluate the expression levels of 584 degradome associated-genes and 117 protease inhibitors (represented in the degradome database http://degradome.uniovi.es/dindex.html), we searched for their expression levels in our microarray data set.
Assessment of protein expression on tissue microarray
Genes that significantly altered their expression level between normal and tumor tissues were evaluated employing protein expression. We made the analysis based on Human Protein Atlas database (HPA), which is a public accessible database containing images that show the spatial distribution of proteins in tissue microarrays by immunohistochemical staining (IHC). 21 The generated tissue microarrays included samples from 46 different normal tissue types from 138 individuals and 20 different kinds of cancer from 216 patients (http://www.proteinatlas.org/). Hence, selected genes were searched for their protein expression on cervical tissue microarray samples. We examined the expression levels of PSMB2, CAST, Cathepsin B (CTSB), USP50, and CST9 proteins on HPA database. The selected antibodies reported on http://www.proteinatlas.org/ were HPA026324 for PSMB2, CAB009491 for CAST, CAB000457 for CTSB, HPA039470 for USP50, and HPA029190 for CST9. Protein expression analyzed on cervical tissue samples (normal cervical epithelia and CC tissues) was reported according to the intensity of immune staining (negative, weak, moderate, and strong); moreover, subcellular localization (nuclear and/or cytoplasmic/membranous) was indicated.
Development of the scoring method
The score of MD risk was evaluated by the development of a mathematical algorithm. This algorithm correlates the level of expression for each gene using the Pearson correlation with values previously established for MD profile and CR profile

where s is the score assigned by the algorithm, PsMDP is the expression profile of the eight gene-degradome signature values corresponding to metastatic group obtained from microarray data, PsCRP is the expression profile of the eight degradome-genes signature with values corresponding to CR group, N is the obtained value for each evaluated gene, and Corr is the Pearson correlation.
The score is obtained by subtracting the correlation from the standard profile for the MD (PsMDP) and the sample (N) to be analyzed, with the correlation of the standard profile CR (PsCRP) and the sample (N) to be analyzed. If the calculated score is greater than zero, the prognosis is metastatic. Conversely, if the score is less than zero, the prognosis is a CR. 18
Disease-free survival analysis
Disease-free survival (DFS) of the resulting patient groups was evaluated using the Kaplan–Meier analysis, and the statistical significance of survival differences was determined with log-rank test (Mantel–Cox) test.
Validation of gene expression profile by quantitative reverse transcriptase polymerase chain reaction
We searched for the most representative genes that could differentiate into metastatic tumors from patients with a complete response. To achieve this objective, we employed the remaining 27 samples to validate the gene expression profile identified in the discovered tumor set. Eight differentially expressed genes (FAM111B, FAM111A, CFB, PSMB9, CASP7, PRSS16, CD74, and PSMB8) were subjected to quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). Each primer set was designed by an experimentally verified computer algorithm and then tested in a quality control assay to guarantee that they yield a single band of the predicted size by agarose gel electrophoresis. The sequence of primers and PCR conditions are shown in Supplemental Table S1. Reverse transcriptase reactions were performed according to the MMLV protocol from Promega (Madison WI, USA) following vendor recommendations. Real-time PCR was performed using FastStart SYBR Green Master. In Light Cycler 480 instrument II (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Duplicate retro-transcribed samples were employed in each assay, and data normalized with β-actin “housekeeping gene.” The comparative Ct method (ΔΔCt) was used to quantify gene expression, and relative quantification was calculated as 2−ΔΔCt.
Results
Clinical characteristics of CC patients
A total of 112 patients from the INCan in Mexico were recruited between April 2010 and August 2012. The range of age varied between 29 and 69 years with a median of 48 years. The majority of patients were classified according to FIGO as IIB (63.3%) and IIIB (22.3%) stages. The squamous cell carcinoma was found in 101 patients (90.1%), while 11 patients were diagnosed with adenocarcinoma (9.8%). As expected, the major HPV types found were type 16 (41%), 18 (19.6%), 45 (11.6%), and others not epidemiologically relevant. The median clinical follow-up was 36 months. A total of 80 patients (71.5b%) were diagnosed as CR during the complete follow-up. In total, 32 patients (28.5%) were diagnosed with MD on new target lesions; the main organs with distal metastases were lungs, and retroperitoneum with 29.4% for each one, followed by liver, and regional lymph with 8.8% for each one. The clinical–pathological characteristics are shown in Table 1.
Expression of degradome-related genes clearly discriminates CC tissues from their normal counterparts
The central mechanism that has to be activated by metastatic cancer cells is the degradation of ECM by the action of a group of proteases that are known as degradome. To obtain a comprehensive picture of degradome-related genes between normal and tumor tissues derived from LACC biopsies, we evaluated the expression levels of the degradome-associated genes and their inhibitors. 22 Our database contained information of 562 proteases grouped into five families (aspartyl, cysteine, serine, threonine, and metalloproteases) and 117 inhibitors (Figure 1). The analysis of the degradome-related gene expression levels in tumor tissues versus normal samples showed two groups of genes differentially expressed between both kinds of samples, which can be observed in Supplementary Figure S1.
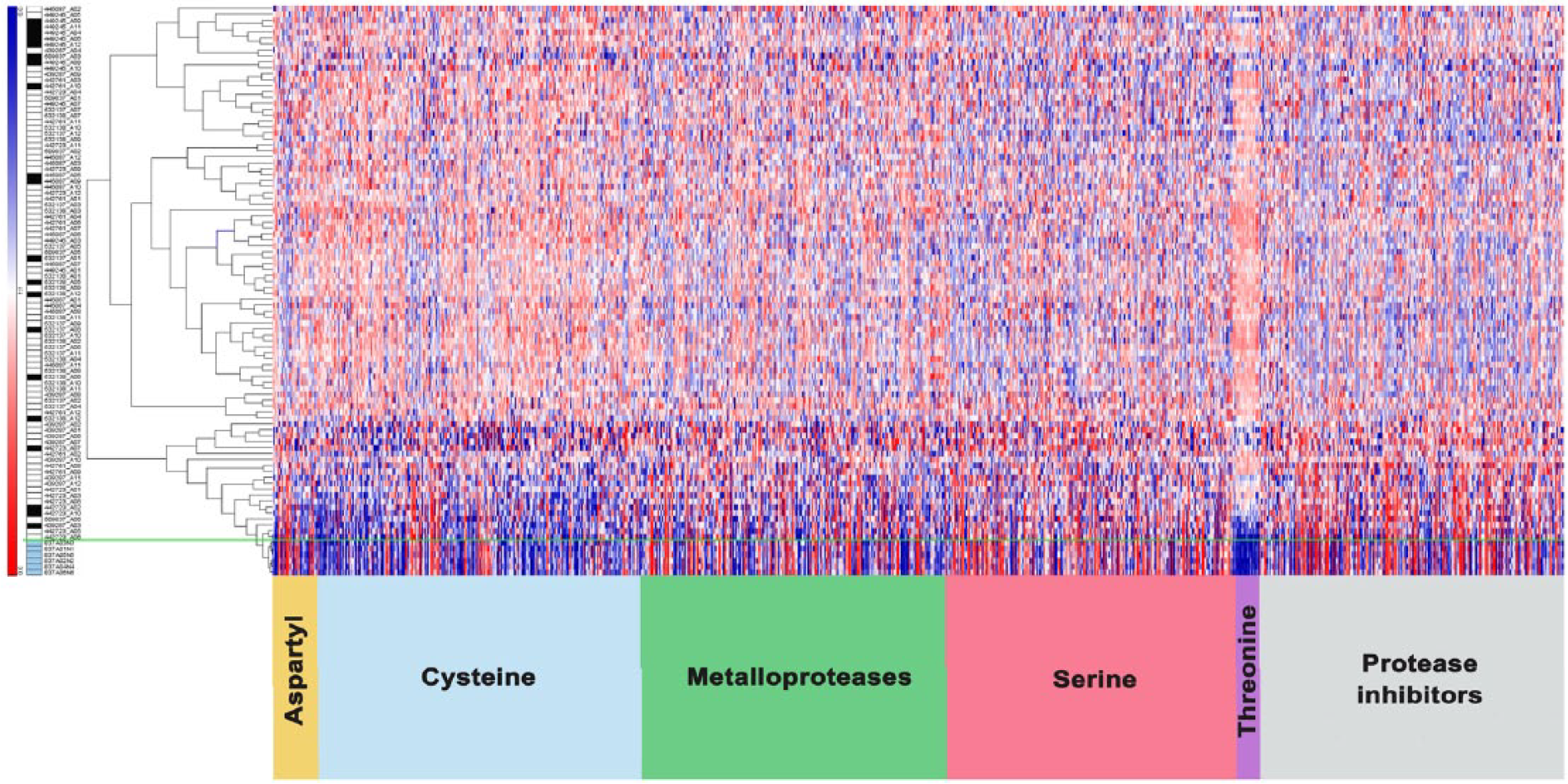
Degradome profile in locally advanced cervical cancer. Global expression profile of the degradome and protease inhibitors through an unsupervised analysis of 701 genes. At the bottom of the image are shown the genes that codify for proteases according to the class they belong to and protease inhibitors. The left column shows the patients with a complete response (white boxes) and the patients with metastasis (black boxes), the normal cervical samples are indicated with the blue boxes.
Using unsupervised clustering, we found that 189 genes belonging to the human degradome are consistently expressed in normal tissues, while 373 genes are under-expressed. Genes that consistently affected their expression between normal and tumor samples included 18 aspartyl proteases (from 23 genes), for example, cathepsin D (CTSD); 122 metalloproteases (from 190), for example, metalloprotease 2 and 9; 118 serine proteases (from 182), for example, tripeptidyl peptidase II (TPP2); 12 threonine proteases (from 15), for example, proteasome subunit alpha; and finally 103 cysteine proteases (from 152), for example, caspases 1, 3, and 8. With respect to inhibitor proteases, 85 of 117 genes, belonging to metalloproteinase (tissue inhibitors of metalloproteinases (TIMPs) 1, 2, 3, and 4), showed altered expression levels in normal and tumor tissues.
Following a significant one-way ANOVA (p < 0.01; Log2 fold change >2), we found that 183 genes showed significant differences between normal and tumor samples, 47 genes were under-expressed in tumor tissue, while 136 were overexpressed. Several proteases found to act as tumor suppressors in other neoplasias. For instance, the Cysteine Proteases USP33 reported as cancer suppressor in colorectal cancer cell 23 or the serine proteases ST14 in colon carcinogenesis. 24 This information allows us to suggest the possibility that the overexpressed proteases of CC could also have tumor-suppressing properties (Supplementary Table S2).
For further confirmation of gene expression levels, we performed an immunohistochemical analysis of normal and cervical tumor sections to evaluate the expression of the top five differential expressed genes supported by the Human Protein Atlas database (http://www.proteinatlas.org/) which has information on protein expression. Thus, in agreement with microarray analysis, positive immunostaining in tumor specimens was observed as follows: PSMB2 (22 positives out of 24 analyzed tissues), CAST (18/22) and CTSB (20/20). On the contrary, the protein expression levels of USP50 (0 of 22 tumor samples analyzed) and CST9 (6 of 24) are low compared to non-tumor tissue (Figure 2). Protein expression showed a dominant cytoplasmic/membranous presence mainly on CC epithelial cells.

Validation of differentially expressed proteins between cervical cancer and normal cervical tissues by immunohistochemistry. Positive immunodetection is observed in brown on the tissue surface. The image shows the expression levels of PSMB2, CAST, and CTSD that were overexpressed in tumor samples; meanwhile, USP50 and CST9 were down-expressed in tumor samples, respecting normal counterparts. These data are consistent with the differential expression of genes obtained by microarray analysis. Bar: 100 µm.
Degradome-related genes are correlated with LACC metastasis
Proteases play a crucial role in the onset and spreading of cancer, mostly in the steps leading to progression from cellular transformation to tumor invasion and metastasis, postulated as a “dependency to proteases” by tumor cells. 25 Such a dependency increases the probability of finding early biomarkers of metastasis by measuring the expression levels of genes encoding proteases and their inhibitors.
Interestingly, to test the hypothesis that degradome-related genes are associated with CC metastases, we correlate clinical data to gene expression profiles that were obtained at the time of diagnosis before patients undergo the conventional treatment. Thus, the expression levels of degradome-related genes were employed to discriminate patients with metastasis or MD and patients who had a CR to treatment (36 months of median follow-up). After ANOVA test, we obtained eight degradome-related genes (FAM111B, FAM111A, CFB, PSMB8, PSMB9, CASP7, PRSS16, and CD74; p < 0.001) (Figure 3).

Eight degradome-related genes are able to distinguish between MD and CR patients. (a and b) Comparison between data derived from microarray and qPCR, respectively. (c) Hierarchical clustering of eight degradome gene levels corresponding to CR (white squares) and MD (black squares) patients. (d) CAT scan at the time of diagnosis (up-left) and 10 months of follow-up. Patients develop metastasis to liver; results were confirmed by PET.
Progression-free survival analysis
Applying the correlation algorithm described in the “Materials and methods” section, we showed that the MD group (positive score) is clearly separated from the CR group (negative score) 4 months after completion of treatment. These results revealed the ability of the eight degradome genes to discriminate patients who develop MD. The survival curve was performed using the log-rank test (Mantel–Cox; p = 0.0002) (Figure 4) (Supplementary Table S3).

Progression-free survival analysis. Kaplan–Meier analysis shows the data of metastasis-free survival and the correlation with the molecular signature predictive of metastasis. Negative values are correlated with a good prognosis, while a positive score with bad prognosis. The survival curve was calculated using the log-rank test (Mantel–Cox; p = 0.0002).
To validate these results in an independent group of 27 patients, we performed an analysis by qRT-PCR of eight genes: FAM111B, FAM111A, CFB, PSMB8, PSMB9, CASP7, PRSS16, and CD74. Supplementary Figure 2 displays scatter plots of 2e−ΔΔCT values obtained from CTs of each eight tested genes in the 27 samples. All graphs show the expression levels of each gene in both groups, CR and MD. Our results are consistent with previous analysis tested in this work, indicating a lower level of expression in MD group compared with CR group, indicating that the molecular signature genes have the ability to classified LACC patients with high risk of metastasis despite being subjected to conventional treatment. For all the above exposed, we propose the molecular signature of degradome as early biomarkers of metastasis.
Discussion
It is well documented that 40% of patients with CC will develop progression or recurrence even through the conventional treatment. Thus, a better understanding of the molecular mechanisms that promote metastasis in CC is a major challenge since the current information about it is controversial and scarce. According to several authors, multiple genes are involved during acquisition of invasiveness and tumor progression. Mainly proteases and associated inhibitors have a fundamental role during different stages of carcinogenesis, mostly in tumor progression and metastasis.9,10,26 Therefore, gene expression microarrays could be a suitable approach to understanding these mechanisms.
With respect to gene expression levels in normal and tumor samples, we found that 183 degradome-related genes were significantly affected in tumor tissues (136 overexpressed and 47 downregulated). For instance, we found that CTSB, which encodes Cathepsin B of the Cysteine proteases family, is implicated in metastatic processes in breast, colon, prostate, and thyroid tumors.27,28 CTSB overexpression is detected in premalignant lesions, suggesting that this enzyme plays an important role during cellular transformation in early stages of malignancy.
Concordantly, we found that metalloproteases MMP-2 and MMP-9 were overexpressed in tumor tissues compared with normal tissues, as reported by Rauvala et al. 29 Such data suggest that these proteases are secreted by tumor cells in LACC patients, explaining in part the metastatic potential of tumor cells. MMPs are a large family of proteases that presents significant changes in CC. In pathological samples, MMPs increase their expression, leading to matrix remodeling, invasion, and metastasis.
Here, we found that 122 MMPs are differentially expressed between normal and cervical tumor tissues. These data establish a correlation with information previously reported, related to the association between increased expression levels of MMPs and tumor progression and invasion processes. 30 Interestingly, we found a series of MMP genes downregulated in tumor tissues (XRCC6BP1, XPNPEP1, SNX3, UQCRC2, TFRC, STAMBP, RNPEPL1 y SLC25A10) (Supplementary Table 2). In contrast, we showed that these genes have high levels of expression in normal tissues, suggesting a possible role as tumor suppressors as described for colorectal carcinoma. 30
In this report, we have discovered a remarkable correlation between distal progression and expression levels of eight degradome-related genes. Little is known about their role in cancer progression and metastasis; furthermore, their potential participation in these biological processes remains to be elucidated. The importance of this article is the association of the eight degradome-related genes to distal progression in CC patients. In a previous report, it was shown that the 11q12 locus is associated with susceptibility to develop prostate cancer, in specific two genes in this region (FAM111A and FAM111B) were correlated with prostate cancer risk. This result suggests that FAM111A and B play a role in tumorigenesis. 31
In gastric adenocarcinoma, nuclear overexpression of PSMB8 is associated with poor prognosis, depth of invasion, lymph node metastasis, and lower survival rates. At the contrary, 32 our results indicate down-expression of PSMB8 correlated to distal progression and metastasis. Moreover, recent studies reported that mir-451 negatively regulates the expression of PSMB8 and suppresses the development and migration of lung cancer. 33 Therefore, PSMB8 participation in the regulation of invasion and metastasis could be associated with specific tumor environment; further functional studies could reveal its role in cancer metastasis. Moreover, PSMB9 has been reported that is downregulated in invasive prostate tumors resistant to castration; concordantly, we were able to show that PSMB9 is downregulated in CC invasive tumors. 34
Here, we have demonstrated the usefulness of detecting LACC patients in risk to develop MD. Although there is no information about the participation of eight degradome-related genes, the expression levels were assessed and confirmed by qRT-PCR in an independent group of CC patients. The most significant contribution of this work was the establishment of a molecular signature derived from degradome genes and its association with clinical response to predict those patients that are at risk of MD.
Footnotes
Acknowledgements
The authors acknowledge Alejandro García-Arriaga, MD, for language editing the manuscript and Nancy Camacho-Ortíz for help as clinical monitor.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
All procedures performed in studies involving human participants were approved by the ethical standards of the institutional and/or national research committee and in accordance with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by Consejo Nacional de Ciencia y Tecnología (CONACyT) (SALUD-2014-233733) and INCan basic research funds. F.R.J. was recipient of COMECyT scholarship 12BCD0054-I.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.