
Abstract
OBJECTIVE:
It is crucially important to discover the relationships between genes and microRNAs (miRNAs) in cancer. Thus, we proposed a combined bioinformatics method integrating Pearson’s correlation coefficient (PCC), Lasso, and causal inference method (IDA) to identify the potential miRNA targets for stomach adenocarcinoma (STAD) using Borda count election.
MATERIALS AND METHODS:
Firstly, the ensemble method integrating PCC, IDA, and Lasso was used to predict miRNA targets. Subsequently, to validate the performance ability of this ensemble method, comparisons between verified database and predicted miRNA targets were implemented. Pathway analysis for target genes in the top 1000 miRNA-mRNA interactions was implemented to discover significant pathways. Finally, the top 10 target genes were identified based on predicted times
RESULTS:
The ensemble approach was confirmed to be a feasible method to predict miRNA targets The 527 target genes of the top 1000 miRNA-mRNA interactions were enriched in 21 pathways. Of note, cell adhesion molecules (CAMs) was the most significant one. The top 10 target genes were identified based on predicted times
CONCLUSION:
The combined bioinformatics method integrating PCC, IDA, and Lasso might be a valuable method for miRNA target prediction, and dys-regulated expression of miRNAs and their potential targets might be prominently involved in the pathogenesis of STAD.
Introduction
Stomach adenocarcinoma (STAD) remains the second largest cause of cancer deaths in the world, which occupies 95% of the gastric malignant tumor [1, 2]. Sadly, STAD is characterized by poor response to chemotherapeutics with a 5-year survival below 20% [3, 4]. The poor outcomes might be attributed to lack of effective strategies for early detection, limited effect of surgery treatment in advanced stages, and weak prognosis of histological indicators. Hence, it is urgent to have a better understanding of the molecular signature of STAD as a critical step towards understanding the pathogenesis and improving our presently limited treatment approaches [5].
Among various regulations of cancer-related genes and pathways in several stages, the regulation of genes by microRNAs (miRNAs) has drawn particular attention, since many miRNAs are situated on chromosomal regions that are frequently changed in cancer [6]. MiRNAs, as a kind of small, non-coding RNA molecules, are highly conserved across species and exert important functions as regulators of gene expression, which have been estimated to regulate as much as 60% of the human protein coding genes [7]. They modulate the levels of targeted genes involved in most biological processes, including development, cell proliferation, apoptosis and differentiation [8, 9, 10]. MiRNAs are aberrantly regulated in cancers, indicating a role as a novel class of oncogenes and tumor suppressors [11]. As a consequence, identifying miRNA functions aids to elaborate the mechanisms underlying STAD and contributes to the design of drugs for effective treatments.
Recently, several computational methods have been created to identify miRNA targets using sequence data, or incorporate expression data into miRNA-mRNA regulatory network to study their relationship [12, 13, 14]. However, the results predicted using different databases or different methods are often inconsistent [15, 16]. Each method usually has a series of hypothesis on data when establishing the model. These assumptions may be suitable for some datasets but may be not fit for the others. Thus, these approaches may not perform well when the underlying relationships violate from these assumptions. It is more urgent to develop more available methods for miRNA target prediction. As documented, ensemble methods which combine different methods perform better than all the individual component methods [16]. Different miRNA target prediction approaches have their own benefits and drawbacks, and their results may be complement. Accordingly, integrating the predictions of multiple methods would improve the prediction performance. Moreover, Le and colleagues have indicated that ensemble methods receive more reliable results than existing individual methods [17].
In the current study, to enhance the understanding of miRNA-mRNA relationship, we proposed a framework that integrated Pearson’s correlation coefficient (PCC), Lasso, and causal inference method (IDA) for predicting miRNA targets based on Borda count election. Using PCC, Lasso, and IDA methods, miRNA target were respectively predicted. Next, using Borda count election, integration of the top 100 predicted targets of each miRNA generated by individual methods were obtained, following by the validation of miRNA target predictions based on the online databases such as Tarbase v6.0, miRecords v2013, miRWalk v2.0, and miRTarBase. Then, pathway enrichment analysis of target genes in the top 1000 miRNA-mRNA interactions was conducted to focus on significant KEGG pathways. These findings provide a better understanding of the mechanism of the initiation and progression of STAD.
Materials and methods
Data set
We collected mRNA expression and miRNA expression data sets for STAD from TCGA. Only samples which simultaneously existed in the two expression sets were reserved for downstream analysis. In the current study, a total of 405 samples were collected. Prior to analysis, the expression data of mRNAs and miRNAs were normalized and log2 transformed after replacing the values equal to zero with the minimum non-null value. After treatment, we obtained 867 miRNAs and 20,262 mRNAs. We then calculated the PCC of expression level for miRNA and mRNA in all samples. The absolute value of PCC for an miRNA-mRNA interaction was defined as
MiRNA target prediction
In the current study, an ensemble method was utilized to predict the miRNA targets, which integrated the three commonly used approaches including PCC, IDA, and Lasso, and took the advantages of the individual method. Thus, to begin with, we presented the specific information of three approaches (PCC, IDA, Lasso) for predicting miRNA target genes based on miRNA and mRNA expression data.
PCC method
PCC is the frequently used index for evaluating the association strength between two variables. With regards to miRNA target prediction, the PCCs in the expression levels of miRNA and mRNA were calculated. Then, miRNA-mRNA pairs were ordered according to the PCC values. Nevertheless, only linear associations are suitable for PCC algorithm, but the associations are non-linear, the effectiveness of PCC method is greatly decreased [18].
IDA method
In anther aspect, IDA introduced by Maathuis et al. [19], evaluates the causal effect that a variable has on the other. Excitedly, Le and colleagues [20] used IDA method to gene expression data to deduce the regulatory relationships between miRNAs and mRNAs, and found that the identified miRNA-mRNA causal regulatory relationships were observed to have a large portion of overlap with the findings of the subsequent gene knockdown experiments. Therefore, this method was covered in our study.
Lasso
Similar to PCC algorithm, Lasso also presented a linear expression correlation of miRNA-mRNA. In the current analysis, for each mRNA, Lasso regression was conducted on all the miRNAs by means of R package glmnet [21]. Similarly, this approach was also included in our analysis.
Ensemble methods of miRNA target prediction
Ensemble methods are planned to take advantage of pros of each individual methods, and to compensate for their cons. Significantly, it has been verified that Borda count election method is a simple but effective strategy to integrate the results from different individual methods, which is a method to choose candidates in a democratic election via selecting the candidate that has the best average rank. In our analysis, Borda count election method was utilized to form ensemble methods. The specific steps of the ensemble methods was shown in the following:
For each miRNA, each of the individual methods (Pearson, IDA, and Lasso) was used respectively to generate the orderings of the mRNAs (as the predicted targets of the miRNA). Apply Borda rank election method to the ordering from step 1 for each miRNA to form a single ranking list of selected mRNAs with respect to the miRNA. Subsequently, the top 100 ranked genes from the list were extracted as the final output, that was to say, these gene were considered as the potential target genes for the given miRNA.
Of note, Borda rank election method is a single but efficient method to combine the results from different separated approaches [22]. Borda rank election method is used to select candidates in a democratic election by choosing the candidate that has the best average rank. Thus, we calculated the average points of the candidate among all representatives, and defined it as the correlation score. The higher the correlation score was, the more significant the predictions were. We sorted the predicted miRNA targets based on their correlation scores, and then we obtained the top
Because the targets of miRNAs validated by experiments still remain limited and there is no complete ground-truth to assess and compare different computational methods, it is a challenge to verify the computation findings [23]. Tarbase, miRecords, and miRTarBase contain confirmed interactions which are manually curated from the literature, and miRWalk focuses on both the predicted and the experimentally validated interactions. In our analysis, Tarbase v6.0 [24], miRecords v2013 [25], miRWalk v2.0 [26], and miRTarBase v4.5 [27] were used to verify the predictions of miRNA targets obtained from the ensemble method.
In-depth analyses of miRNA potential targets
Top 50 miRNA-mRNA interactions. Red nodes were miRNAs and blue nodes denoted mRNAs. The edges stood for the interactions between any two of miRNA and mRNA.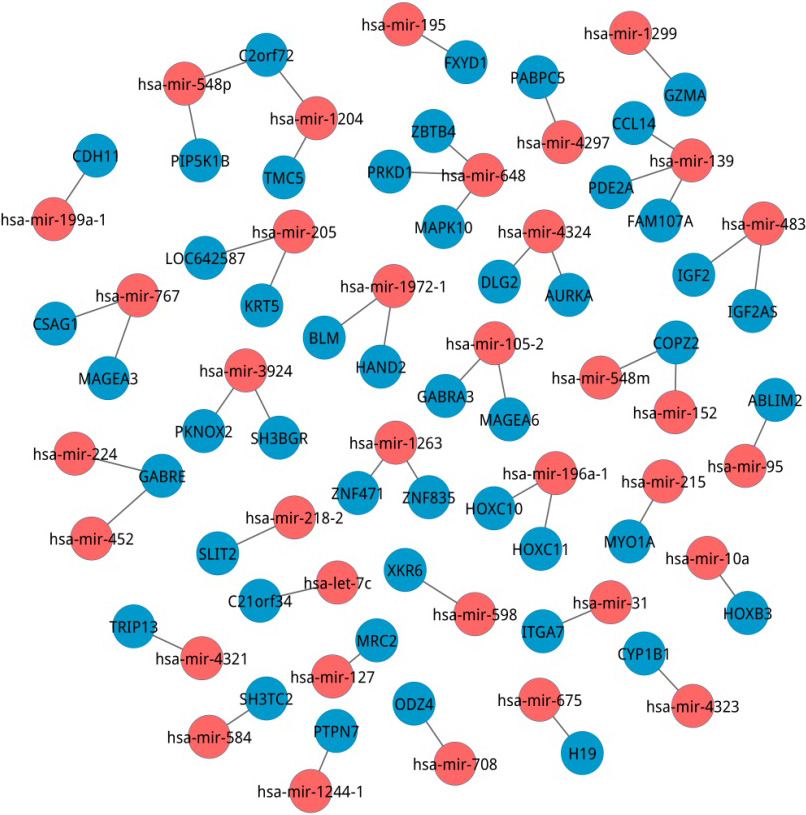
With regard to functional analysis of miRNA potential targets in the top
It is well known that a miRNA can regulate multiple genes [28], and a gene can be targeted by several miRNAs [13]. Changes in these relationships can alter the biological functions associated with a specific tumor [29]. Thus, the gene which was predicted by more times or which was targeted by more miRNAs, perhaps was more significant than those only was predicted by one time. For this reason, the predicted occurrence frequency for mRNAs among
MiRNA targets prediction
The top 50 highly-confident novel miRNA-mRNA interactions in the stomach adenocarcinoma (STAD)
The top 50 highly-confident novel miRNA-mRNA interactions in the stomach adenocarcinoma (STAD)
A total of 21 pathways of 527 target genes in top 1000 miRNA-mRNA interactions based on false discovery rate (FDR)
In the current work, based on the expression data of STAD downloaded from the TCGA database, 691 mRNAs and 85 miRNAs were reserved for further analysis after filtration treatments. An ensemble method integrating three approaches (Pearson
The occurrence frequency of the top 10 target genes of miRNAs
The top 50 predicted miRNA-mRNA interactions were listed in Table 1, and From the Table 1, we found that the top 19 miRNA-mRNA interactions had the same correlation scores and were determined as the most significant interactions. For example, PTPN7 had the correlation score of 691 and its miRNA was miRNA-1244-1, BLM (score
There are 20095 interactions among 228 miRNAs, 21590 interactions with 195 miRNAs, 1710 interactions with 226 miRNAs, and 37372 interactions with 576 miRNAs for Tarbase, miRecords, miRWalk, and miRTarBase databases, respectively. After eliminating the duplicates, 62,858 unique interactions were kept as background interactions to validate the predicted results in our study. Taking intersections of all predicted miRNA-mRNA interactions and background interactions, 62 intersected interactions were extracted, which further indicated that the ensemble method was a available and valuable method for predicting miRNA targets.
In-depth analyses of miRNA targets
KEGG pathway enriched analysis for 527 target genes of the top 1000 miRNA-mRNA interactions was implemented. Based on the significance set as FDR
The gene which was predicted by more times or which was regulated by more miRNAs, perhaps was more significant than those only was predicted by one time. This might provide another way to assess the significance of one gene in a certain cancer. Thus, the prediction frequency for mRNAs was compared, and the targets with the predicted times
Discussion
MiRNAs are a class of non-coding RNA molecules which are widely expressed in human tissues with significant power to mediate several biological activities [30]. In this condition, miRNAs provide a way to elaborate the complex mechanisms within the disease status, including cancer. In our study, to enhance the understanding of miRNA functions, we proposed a combined bioinformatic approach integrating PCC, Lasso, and IDA to the identification of miRNA targets based on Borda count election. The 527 target genes of the top 1000 miRNA-mRNA interactions was enriched in 21 pathways. Of note, the pathway of cell adhesion molecules (CAMs) was the most significant one, which had the maximum number of target genes, but owned the smallest FDR value. Moreover, all the top 10 target genes were targeted by 4 miRNAs, and target genes GABRA3 and CSAG1 were simultaneously targeted by miRNA-105-1, miRNA-105-2, and miRNA-767. Significantly, among the top 19 miRNA-mRNA interactions with the highest correlation scores (score
As documented, cell adhesion is one important step in cancer metastasis. Syndecan-1, cadherin and integrin consist of cell adhesion molecules (CAMs) [31]. Clearly, CAMs participate in a broad range of biological processes, such as cell-cell and cell-matrix interactions, cell cycle, cell migration, and signaling during development and tissue regeneration [32]. Nevertheless, aberrant expression of CAMs disrupts normal cell-cell and cell-matrix interactions, freeing cells from normal check points and constraints, and facilitating tumor formation and metastasis [33]. Expression of the CAMs has been demonstrated to be related with many cancers, including breast cancer [34], clear cell renal carcinoma [35], gastric carcinoma [36], and colorectal cancer [37]. Accordingly, the pathway of cell adhesion molecules (CAMs) might play important roles in the progression of STAD.
Moreover, in our study, among the top 10 target genes, GABRA3 and CSAG1 were simultaneously targeted by miRNA-105-1, miRNA-105-2, and miRNA-767. GABRA is one kind of GABA receptors, and GABRA is ligand-gated chloride channels made up of five subunits which are encoded by 19 different genes that have been grouped into eight subclasses based on sequence homology (
In addition to GABRA3, PTPN7/miRNA-1244-1 had the highest correlation score of 691 among the top 50 highly-confident miRNA-mRNA interactions predicted by ensemble method, and PTPN7 was one of the top 10 targets in our study. PTPN7 belongs to a class of PTP family containing a group of dual-specificity phosphatases (DUSPs) which regulate MAPKs activity [51]. MAPK plays a critical role in the mediation of inflammatory, oncogenic signals and gastric adenocarcinoma [52]. Moreover, it is known that the MAPK pathway is involved in the induction of MMP-2, which has been implicated to be related with the progression and metastasis of gastric carcinoma [53]. Significantly, Wu et al. [54] have indicated that PTPN7 play important roles in STAD differentiation and progression. In our study, miRNA-1244-1 was the corresponding miRNA. MiRNA-1244-1 is one of three genomic locis encoding miRNA-1244. Significantly, miRNA-1244 has been demonstrated to play important roles in the differentiation of gastric cardia adenocarcinoma [55]. Of note, miRNA-1244-1 has been implicated to be associated with regulation of cell proliferation [56]. Moreover, regulation of cell proliferation was found to be connected with MMPs [57]. Thus, based on these results, we infer that miRNA-1244-1 expression may be not only promising therapeutic targets but also serve as valuable prognostic markers for STAD, via regulating the expression of PTPN7.
In summary, these findings indicated that the combined bioinformatics method which integrated PCC, IDA, and Lasso might be a valuable method for miRNA target prediction, and also suggested dys-regulated expression of miRNAs and their potential target mRNAs were prominently involved in the pathogenesis of STAD. However, subsequent validation of these observations in cell culture confirmed that GABRA3 and CSAG1 are both regulated by miRNA-105-1, miRNA-105-2, and miRNA-767 in STAD cells. Moreover, it remains to be investigated whether the high-confident novel miRNA targets found in our study also participate in the progression of STAD. In addition, correlation analysis of microRNA and mRNA expression in human tissue may prove useful in identifying mRNAs that are regulated by miRNAs.