
Abstract
Background:
This study aimed to uncover potential microRNA (miRNA) targets in head and neck squamous cell carcinoma (HNSCC) using an ensemble method which combined 3 different methods: Pearson’s correlation coefficient (PCC), Lasso and a causal inference method (i.e., intervention calculus when the directed acyclic graph (DAG) is absent [IDA]), based on Borda count election.
Methods:
The Borda count election method was used to integrate the top 100 predicted targets of each miRNA generated by individual methods. Afterwards, to validate the performance ability of our method, we checked the TarBase v6.0, miRecords v2013, miRWalk v2.0 and miRTarBase v4.5 databases to validate predictions for miRNAs. Pathway enrichment analysis of target genes in the top 1,000 miRNA–messenger RNA (mRNA) interactions was conducted to focus on significant KEGG pathways. Finally, we extracted target genes based on occurrence frequency ≥3.
Results:
Based on an absolute value of PCC >0.7, we found 33 miRNAs and 288 mRNAs for further analysis. We extracted 10 target genes with predicted frequencies not less than 3. The target gene MYO5C possessed the highest frequency, which was predicted by 7 different miRNAs. Significantly, a total of 8 pathways were identified; the pathways of cytokine–cytokine receptor interaction and chemokine signaling pathway were the most significant.
Conclusions:
We successfully predicted target genes and pathways for HNSCC relying on miRNA expression data, mRNA expression profile, an ensemble method and pathway information. Our results may offer new information for the diagnosis and estimation of the prognosis of HNSCC.
Keywords
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the 6 most common malignancies globally, with an incidence of about 600,000 diagnosed patients per year (1). The main risk factors for HNSCC include consumption of tobacco and alcohol, as well as human papillomavirus infection (2, 3). The treatment for HNSCC covers surgery with or without concurrent chemotherapy and radiation therapy, or radiation alone. Despite employing these advanced techniques and efficient chemotherapy drugs, the overall mortality rate remains at approximately 50%, partly because of advanced disease stage at diagnosis and the relatively high recurrence rates (4, 5). Thus, there is an urgent need to identify reliable biomarkers and develop more effective therapeutic agents to further improve patient prognoses.
In recent years, more attempts have been made to detect genomic alterations which promote the occurrence and progression of cancer, but most efforts have focused on genes (6, 7). Significantly, these gene biomarkers have had limited success in the clinical setting. Currently, the knowledge of noncoding genes – e.g., microRNA (miRNA) contributing to the initiation and propagation HNSCC – is limited. MiRNAs, as a class of important endogenous small noncoding RNAs, control gene expression via promoting messenger RNA (mRNA) degradation and inhibiting translation (8). They participate in many important cancer-related processes including proliferation (9, 10), differentiation (11), metabolism (12), apoptosis (13) and even cancer development and progression (14). Moreover, the link between miRNAs and cancers demonstrates that these small RNAs can function as therapeutic agents (15). Thus, identification of miRNA functions is helpful for illuminating the molecular mechanisms of HNSCC and aiding in drug design for treatment.
Recently, many computational methods have been developed to extract miRNA targets (16-18). Unfortunately, the results predicted using different approaches are frequently conflicting (19, 20). Thus, developing more reliable methods for predicting miRNA targets is urgent. It has been reported that ensemble methods integrating different approaches perform better than any of the single-component methods (19). Different miRNA target prediction methods have their own benefits and limitations, and this combined approach can solve the problem of inconsistent results. In addition, as Le et al (21) have demonstrated, ensemble methods obtain more reliable results than existing individual approaches.
In the current study, we aimed to identify the miRNA targets related to HNSCC, using ensemble methods. Specifically, we downloaded the files of miRNAs and mRNAs of HNSCC from the Cancer Genome Atlas (TCGA) database. Then, Pearson’s correlation coefficient (PCC), Lasso and a causal inference method (i.e., intervention calculus when the directed acyclic graph (DAG) is absent [IDA]) were used for miRNA target prediction. Next, the Borda count election method was used to integrate the top 100 predicted targets of each miRNA generated by individual methods. Afterwards, we utilized 4 databases, TarBase v6.0 (22), miRecords v2013 (23), miRWalk v2.0 (24) and miRTarBase (25), to validate the predictions for miRNAs. Then, pathway enrichment analysis of target genes in the top 1,000 miRNA–mRNA interactions was conducted to focus on significant KEGG pathways. Finally, we extracted the target genes based on an occurrence frequency ≥3. The flow diagram of this method is shown in Figure 1.
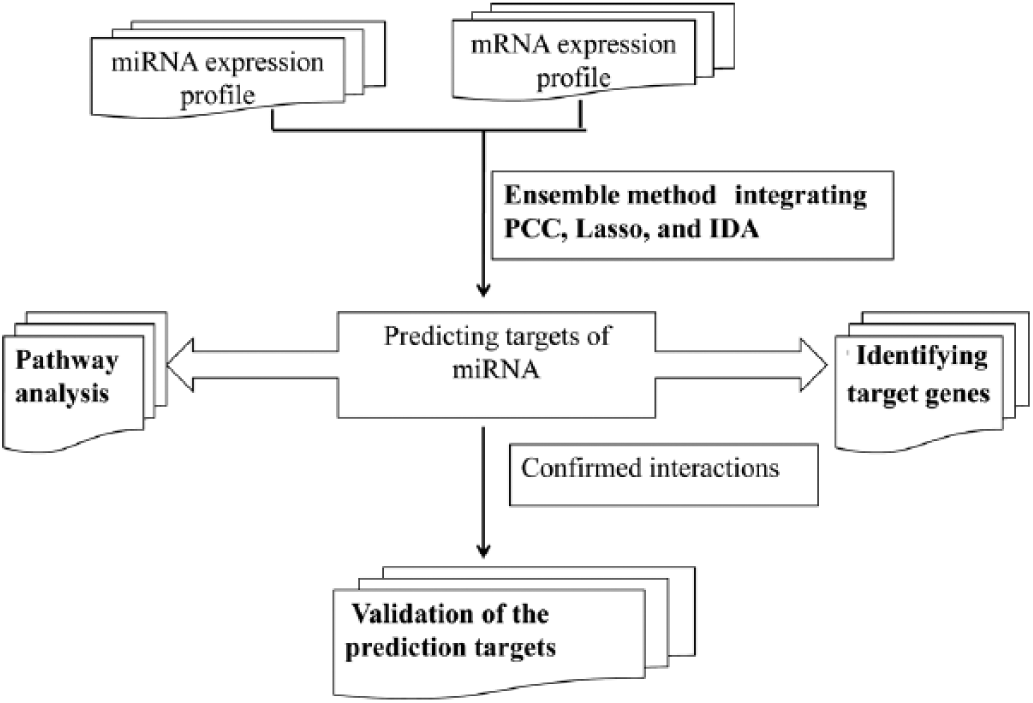
Flow diagram of this method. IDA = intervention calculus when the directed acyclic graph is absent; miRNA = microRNA; mRNA = messenger RNA; PCC = Pearson’s correlation coefficient.
Materials and methods
Microarray data
The TCGA database is a comprehensive and coordinated effort to accelerate our understanding of the molecular basis of cancer by providing a huge amount of genomic data obtained with the ultimate genome analysis technologies. In our study, miRNA and mRNA expression profiles of HNSCC were obtained from the TCGA database (http://cancergenome.nih.gov/). Following data filtering, only samples with both miRNA expression profile and mRNA expression profile were kept for subsequent analysis. Ultimately, a total of 524 cases were obtained.
Prior to analysis, with the goal of controlling the quality of mRNA and miRNA in the samples, we retreated the raw data. Specifically, miRNAs or mRNAs whose expression values were equal to 0 in a sample were discarded. Then, raw signal data were first transformed to a log2 scale, followed by global variance stabilization normalization (26) of all of the arrays. After pretreatment, a total of 903 miRNAs and 20,231 mRNAs remained. After that, to compute miRNA–mRNA interaction weights, the PCC distribution of the miRNA–mRNA in all samples was calculated to measure the correlation strength between miRNA and mRNA. The absolute value of PCC for an miRNA–mRNA interaction was defined as δ. The higher the weight value was, the stronger the interaction was. In our study, only miRNA–mRNA interactions whose correlations were higher than the predefined threshold δ (δ = 0.7) were reserved for subsequent analysis.
Methods for predicting miRNA targets
In this stage, we first introduced 3 commonly used approaches (PCC, IDA and Lasso) for miRNA target prediction based on gene expression information.
PCC method
PCC is the commonly used method for evaluating the association strength between a pair of variables. For miRNA target prediction, the PCCs in the expression levels between miRNA and mRNA were computed. The miRNA–mRNA pairs were then sorted based on the PCC values. Because miRNAs mainly down-regulate the expression of mRNAs, the negative correlations were ordered at the top of the ranking list to favor the down-regulation according to the absolute values. Unfortunately, PCC is projected to obtain only linear associations, and its effectiveness is greatly decreased when the associations are nonlinear (27).
IDA method
The IDA method, as a causal inference method introduced by Maathuis et al (28), is used to evaluate the causal effect that a variable has on the other. Recently, Le and coworkers (29) have applied IDA to gene expression datasets to deduce the regulatory relationships between miRNAs and mRNAs. The identified miRNA–mRNA causal regulatory relationships were observed to have a large portion of overlap with the findings of the subsequent gene knockdown experiments. Thus, this method was included in our study.
Lasso
Lasso is a high-dimensional regression technique which can be employed to infer the associations between variables. In the current analysis, the expression level of each mRNA was exhibited as a linear expression of the expression levels of all miRNAs, and the coefficient of a miRNA in the regression model was applied as the association strength between miRNA and mRNA. For each mRNA, Lasso regression was performed on all of the miRNAs using the R package Glmnet (30). Similar to the PCC method, the miRNA–mRNA pairs that had negative effects were sorted at the top to favor the down-regulation. Likewise, we also included this approach in our analysis.
Ensemble methods of miRNA target prediction
Ensemble methods make use of individual methods, in an attempt to compensate for their disadvantages. In detail, individual methods are used to order the miRNA–mRNA interactions on the basis of some criteria including the strength of the correlation coefficients, and ensemble methods integrate the orderings from different methods to produce a new sorting. So far, different strategies for integrating different rankings have been developed, such as taking the intersection, the union and the average of the top k results predicted by individual methods. Significantly, the Borda count election method has been verified to be a simple but effective strategy for combining the results from different individual methods. It is a method to choose candidates in a democratic election via selecting the candidate that has the best average rank. Of note, using the Borda average rank of multiple methods does not lead to a method that averages the performance of the individual methods. Actually, we aimed to design a method that was superior to the individual methods. In our paper, we utilized the Borda count election method to form ensemble methods. Thus, we did not employ the whole sorting list generated by each individual method to form the ordering for the ensemble approaches, but only applied the top 100 predicted targets of an miRNA generated by each method.
Ground truth for verification
It is difficult to verify the computation findings, since the counting of experimentally validated targets of miRNAs is still limited, and there is no complete ground truth for assessing and comparing different computational methods (31). Fortunately, TarBase and miRecords, as well as the miRTarBase database contain confirmed interactions which are manually curated from the literature, and miRWalk includes both the predicted and the experimentally validated interactions. In the present analysis, we utilized the 4 databases, TarBase v6.0 (22), miRecords v2013 (23), miRWalk v2.0 (24) and miRTarBase (25), to verify the predictions of the methods for all miRNAs. There were 20,095 interactions among 228 miRNAs, 21,590 interactions with 195 miRNAs, 1,710 interactions with 226 miRNAs and 37,372 interactions with 576 miRNAs for the TarBase, miRecords, miRWalk and miRTarBase databases, respectively,. After removing the duplicates, a total of 62,858 unique interactions remained for validation, which were defined as confirmed interactions in our study.
Functional analyses of target genes
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was applied to explore the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the target genes in the top ω miRNA–mRNA interactions (32). To correct the p values for the multiple tests, the false discovery rate (FDR) was estimated (33). An FDR <0.05 and gene count >5 were regarded as the cutoff criterion.
Discovery of occurrence frequency of predicted target genes
Usually, an miRNA can target multiple genes, and several miRNAs also regulate the expression of the same gene. Given this possibility, we predicted the target genes for the top ω miRNA–mRNA interactions, and then we analyzed the frequency of these predicted target genes. Next, the target genes were sorted based on their occurrence frequency. Finally, we obtained the target genes based on an occurrence frequency ≥3.
Results
Predicting miRNA targets
Using the expression data for HNSCC in the TCGA database, a total of 33 miRNAs and 288 mRNAs remained for subsequent analysis after pretreatment. After integrating 3 approaches (Pearson + Lasso + IDA) based on the Borda count election, an ensemble method was obtained, and then we applied this ensemble method to perform miRNA target predictions. Based on miRNA–mRNA interactions, the miRNA targets (named as genes or mRNAs) were extracted. In this prediction process, a correlation score was assigned to each miRNA–mRNA interaction, and all miRNA–mRNA interactions were sorted in descending order. The greater the correlation score, the more meaningful the prediction results were. Because there were a large number of miRNA targets, we only selected the top ω (ω = 1,000) interactions as study objects.
The top 50 predicted miRNA–mRNA interactions are shown in Table I. In Table I, we observed that the top 10 miRNA–mRNA interactions had the greater correlation scores and were considered to be the most significant interactions. Specifically, the interaction with the greatest score was that between MYO5C and hsa-mir-1266 (score 288). The others were GABRE and hsa-mir-224 (score 285), HOXA10 and hsa-mir-196b (score 282), IGF2 and hsa-mir-483 (score 281), LOC642587 and hsa-mir-205 (score 273), MGC16121 and hsa-mir-503 (score 270), DHRS7C and hsa-mir-499 (score 256), SH3TC2 and hsa-mir-584 (score 251), TNFRSF17 and hsa-mir-148a (score 237) and XKR6 and hsa-mir-598 (score 229). From Table I, we also found that hsa-mir-1266 regulated 4 genes (MYO5C, KRT16, KRT6B and KRT6C), and KRT6B was simultaneously regulated by hsa-mir-598 and hsa-mir-1266. Thus, an miRNA can regulate a plurality of genes, and some different miRNAs simultaneously regulated the expression of a particular gene.
Top 50 predicted mRNA—miRNA interactions
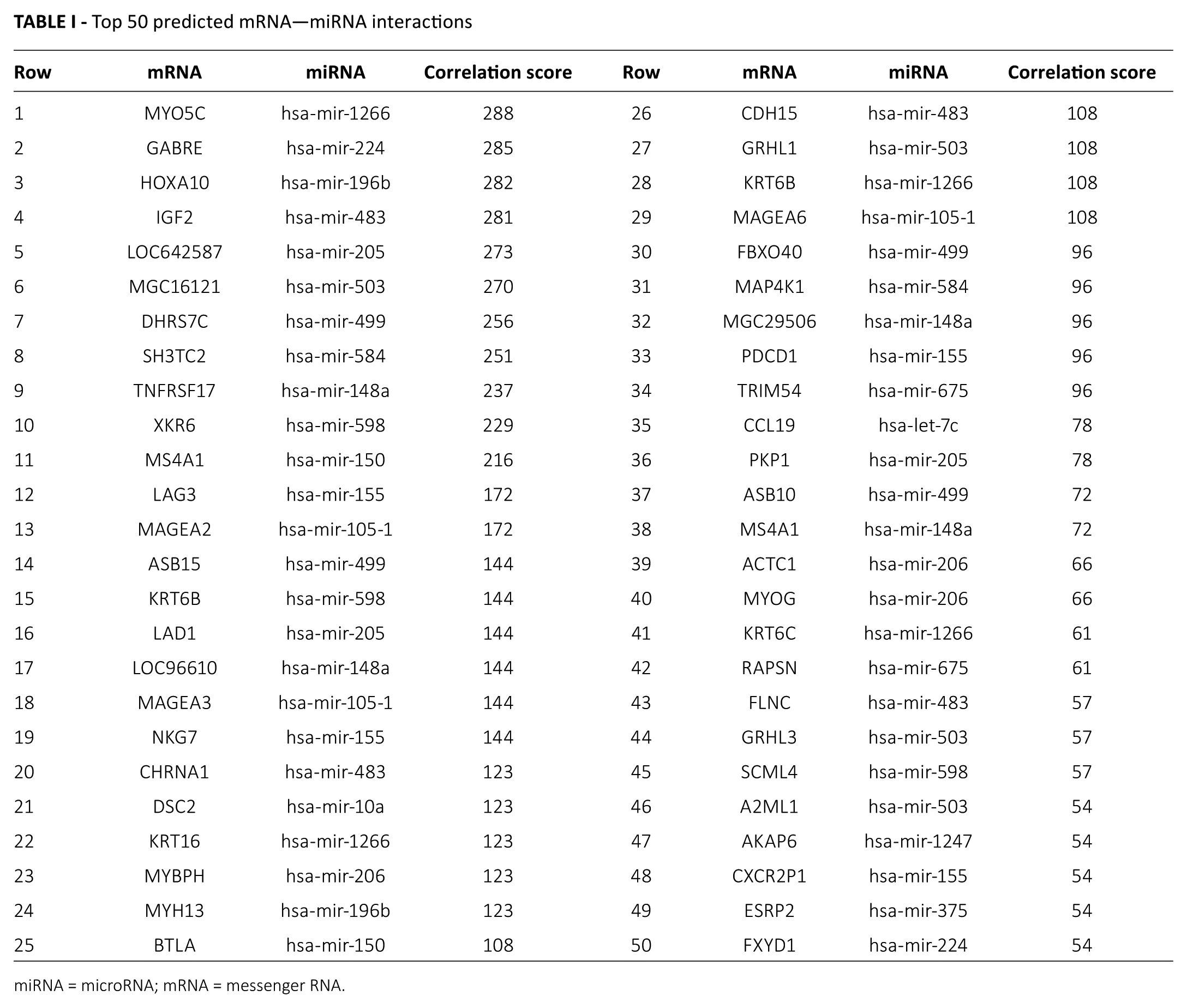
miRNA = microRNA; mRNA = messenger RNA.
Validation of the predicted targets
In an attempt to verify the prediction results for miRNA targets extracted using the ensemble method, the experimentally verified databases miRTarBase, TarBase, miRecords and miRWalk were utilized. After removing duplicates, 62,858 unique interactions remained for validation, which were defined as confirmed interactions in our study. For each miRNA, we selected the top 100 target genes for verification. Significantly, we found that 25 miRNA–mRNA interactions were validated by the experimentally confirmed databases. This result further indicated the feasibility of the ensemble method.
Functional analyses of target genes
To gain a better understanding of the biological pathways of target genes, we used DAVID software to implement the pathway enrichment analyses for 278 target genes in the top 1,000 miRNA–mRNA interactions, with the focus on significant KEGG pathways based on the cutoff of FDR <0.05 and gene count >5. A total of 8 enrichment KEGG pathways were identified; Specific information for these is shown in Table II. Remarkably, 4 out of 8 pathways were signaling-related pathways. Among these, the top 5 pathways were the T-cell-receptor signaling pathway, calcium signaling pathway, cell adhesion molecules (CAMs), cytokine–cytokine receptor interaction and chemokine signaling pathway.
Total of 8 pathways of 278 target genes in the top 1,000 miRNA—mRNA interactions based on FDR <0.05
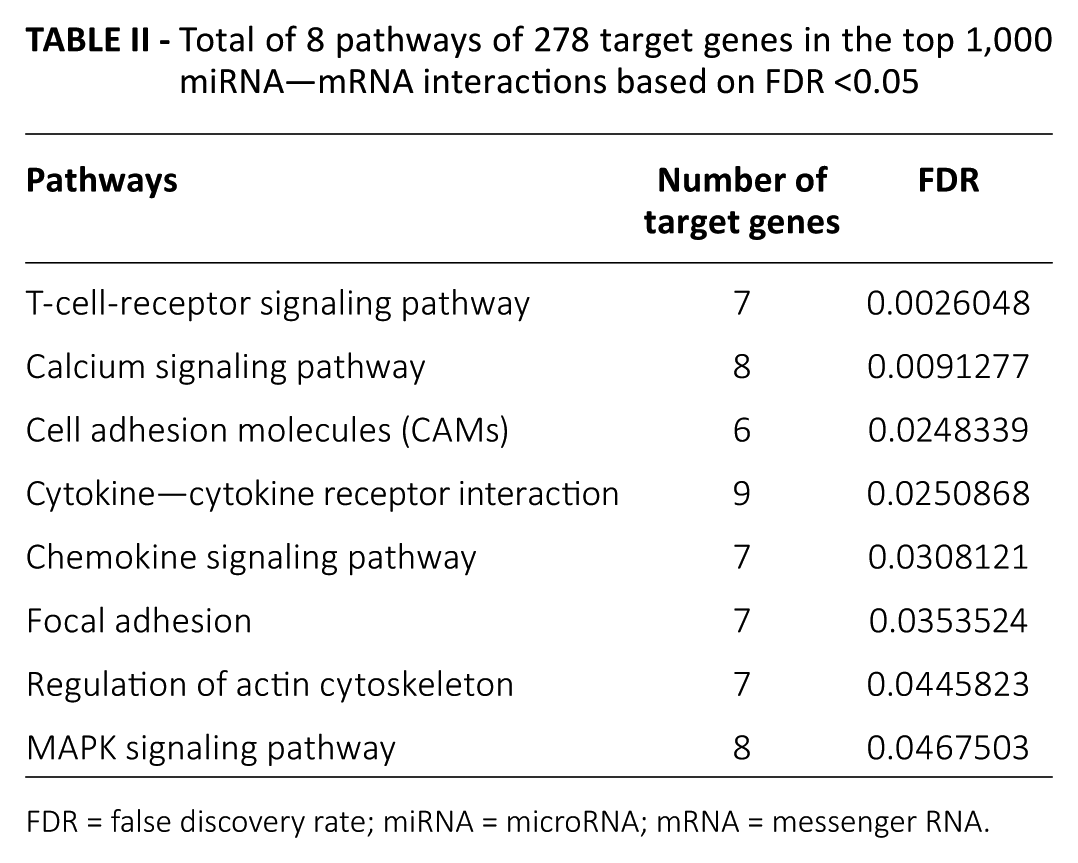
FDR = false discovery rate; miRNA = microRNA; mRNA = messenger RNA.
Occurrence frequency of predicted target genes
Any given gene that was predicted more times or which was regulated by more miRNAs, was probably more significant compared with those only predicted 1 time. To a certain degree, this provides another way to assess the significance of a gene in a specific cancer. Therefore, among the 1,000 miRNA–mRNA interactions, the occurrence frequencies for mRNAs were calculated. The targets having occurrence frequencies >3 are listed in Table III. Based on the occurrence frequencies of the predicted target genes, a total of 10 target genes – MYO5C, FXYD1, PYGM, SYNPO2, CKMT2, PPP1R1A, KRT16, CCL19, HOXA10 and ABRA – were identified, as shown in Table III. In this table, we observe that target gene MYO5C possessed the highest frequency, and was predicted by 7 different miRNAs, and the target genes FXYD1 and PYGM were each regulated by 5 miRNAs simultaneously.
List of target genes of miRNAs, based on occurrence frequency ≥3
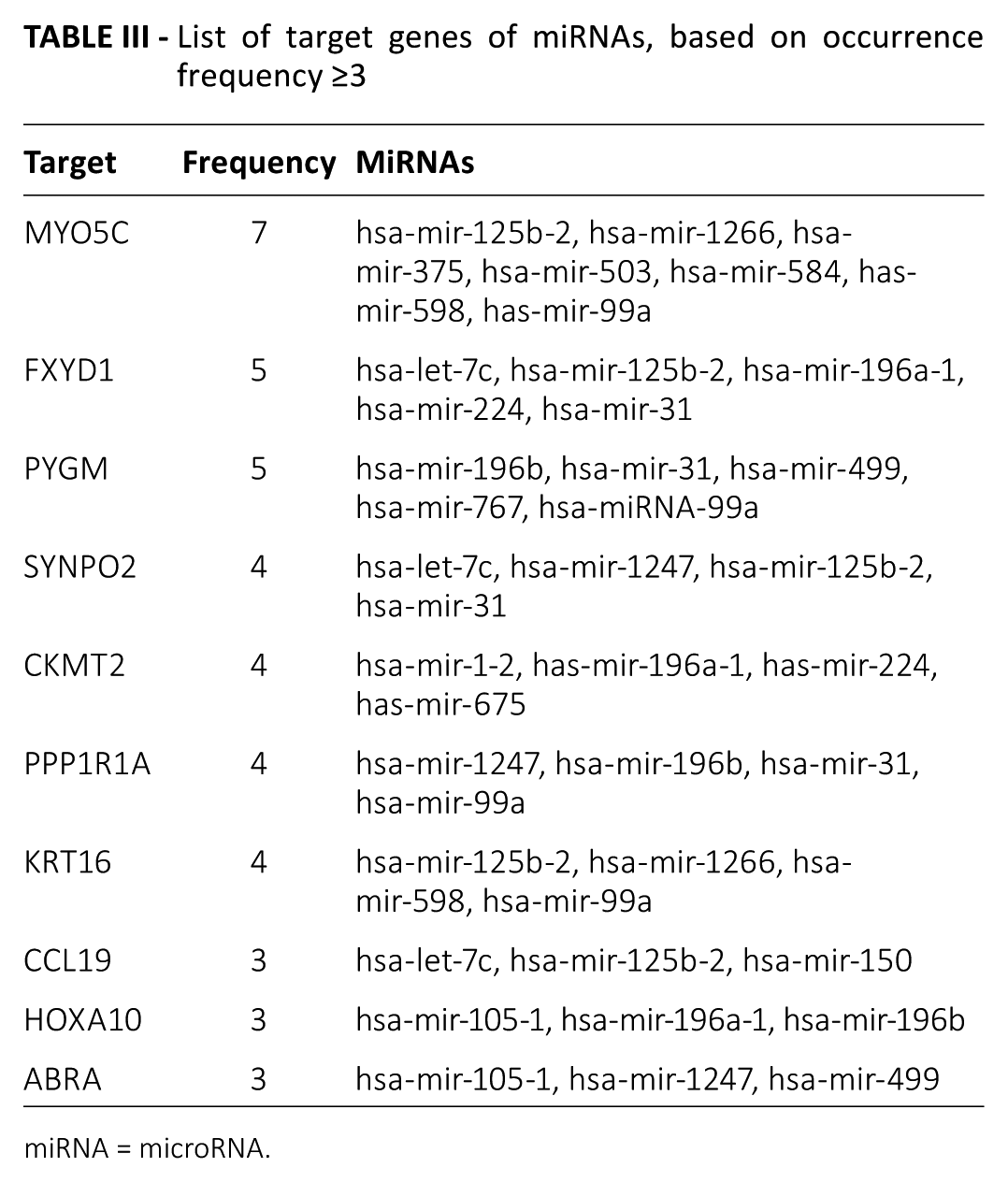
miRNA = microRNA.
Discussion
HNSCC is a common type of nonskin cancer that occurs worldwide (34). New bio-signatures are required to improve predictions and treatment outcomes for HNSCC. Of note is the fact that miRNAs seem to be a good candidate for this disease. Thus, finding miRNA functions is useful to elucidate the molecular mechanisms underlying fatal diseases such as HNSCC. Several computational approaches have been created to identify miRNA targets (35), based on structural stability or sequence complementarity of the putative duplex. Although these methods produce several potential target genes, the findings may contain a high-rate of false negatives and false positives (36). In addition, the results predicted using different methods often lack consistency (36), because miRNA binding sites are small, and therefore they do not support statistically significant predictions (37). Recently, many computational methods have been designed to combine expression profiles with the study of miRNA–mRNA regulatory relationships. It has been reported that computational methods perform inconsistently in inferring gene regulatory networks with different datasets (19). Each method usually has a set of hypotheses regarding the data when building the model. These hypotheses may be suitable for some datasets, yet may be invalidated in others. Hence, it is crucially important to devise more effective methods to predict miRNA targets. As we know, different miRNA target prediction methods have their own advantages and limitations, and their results may complement each other. Thus, combining the predictions of multiple methods might improve the predictive power. Fortunately, ensemble methods integrating different methods perform better than any of the individual component methods (19).
In our study, we introduced a framework based on Borda count election to construct an ensemble method which integrated PCC, Lasso and IDA to identify the miRNA targets, and further verified it via assessing correspondences between predicted miRNA–mRNA interactions and confirmed interactions. Based on this ensemble method, we sorted the predicted miRNA targets for HNSCC patients according to their correlation scores and found the top 1,000 miRNA–mRNA interactions. For the given miRNA–mRNA interactions, the predicted times for 1 target gene were calculated. Significantly, in our study, the target gene MYO5C possessed the greatest frequency, which was predicted by 7 different miRNAs. Specifically, the interaction of MYO5C and hsa-mir-1266 had the highest score (score 288). MYO5C, as a member of the class V myosins, which is the most efficient class in actin-mediated transport, drives the actin-mediated membrane trafficking pathway (38). There is a growing body of evidence that the actin-based processes – e.g., cell polarity and cell migration – may play key roles in advanced tumors and may be associated with their invasion into adjacent tissues (39, 40). As reported, the MYO5C mutation has been observed in colon tumor histologies (41).
A previous study demonstrated that hsa-mir-1266 may promote the progression of gastric cancer, and hsa-mir-1266 is positively associated with a better prognosis for patients with gastric cancer (42). Sevinc et al (43) have suggested that higher hsa-mir-1266 might be a significant prognostic factor for recurrence and/or metastasis in breast cancer. Interestingly, Saad et al (44) found that hsa-mir-1266 exhibited significant overexpression in alcohol-related HNSCC. Additionally, the interaction of MYO5C and hsa-mir-1266 has also been predicted to be one of the significant mRNA–miRNA connections in multiple tumor types based on a correlation coefficient >0.6 (45). Nevertheless, there is little current knowledge about the involvement of MYO5C in HNSCC.
Moreover, no studies have investigated the regulatory mechanisms of MYO5C and has-mir-1266. This is the first time the correlation between MYO5C/has-mir-1266 and HNSCC has been explored. We infer that HNSCC progression might result from the dys-expression of MYO5C/hsa-mir-1266 through actin-based functions, which further influences changes in cellular adhesion and transportation.
To extract the functional gene sets involved in the targets of miRNA, we completed pathway enrichment analysis. Of note, the cytokine–cytokine receptor interaction was one of the most significant pathways. As demonstrated, cytokines, as soluble extracellular proteins, have been indicated to induce inflammatory responses which exert decisive functions in tumor development (46). These responses are triggered via binding to the corresponding receptors of cytokines (47). Significantly, cytokine–cytokine receptor interactions in the cancer milieu have been indicated to play important roles in cancer progression (48).
Another significant pathway in our study was the chemokine signaling pathway. Chemokines are crucial regulators in inflammation responses, and in parallel, members of the chemokine system play a wide variety of roles in cancer occurrence and progression (49, 50). Moreover, metastatic HNSCC expresses a chemokine receptor which activates phosphoinositide-3 kinase to promote invasion and HNSCC (51). Thus, we infer that the cytokine–cytokine receptor interaction pathway and chemokine signaling pathway might contribute to the progression of HNSCC, thereby suggesting that the ensemble method using PCC, Lasso and IDA is a good method for miRNA target prediction.
In spite of the aforementioned results, there remained several limitations in our analysis. The predicted results should be verified by laboratory experiments. In further studies, the expression levels of the high-confidence novel miRNA targets will be confirmed in a large sample of HNSCC patients.
These findings indicate that the ensemble method that integrates PCC, Lasso and IDA, might be a valuable method for miRNA target prediction. Moreover, our results may provide new information for the diagnosis and estimation of the prognosis of HNSCC, and may contribute to further experimental studies.
Footnotes
Disclosures
Financial support: No grants or funding have been received for this study.
Conflict of interest: We declare that no conflict of interest existed.