
Abstract
Abnormal miRNA expression has been evidenced to be directly linked to HCC initiation and progression. This study was designed to detect possible prognostic, diagnostic, and/or therapeutic miRNAs for HCC using computational analysis of miRNAs expression. Methods: miRNA expression datasets meta-analysis was performed using the YM500v2 server to compare miRNA expression in normal and cancerous liver tissues. The most significant differentially regulated miRNAs in our study undergone target gene analysis using the mirWalk tool to obtain their validated and predicted targets. The combinatorial target prediction tool; miRror Suite was used to obtain the commonly regulated target genes. Functional enrichment analysis was performed on the resulting targets using the DAVID tool. A network was constructed based on interactions among microRNAs, their targets, and transcription factors. Hub nodes and gatekeepers were identified using network topological analysis. Further, we performed patient data survival analysis based on low and high expression of identified hubs and gatekeeper nodes, patients were stratified into low and high survival probability groups. Results: Using the meta-analysis option in the YM500v2 server, 34 miRNAs were found to be significantly differentially regulated (P-value ⩽ .05); 5 miRNAs were down-regulated while 29 were up-regulated. The validated and predicted target genes for each miRNA, as well as the combinatorially predicted targets, were obtained. DAVID enrichment analysis resulted in several important cellular functions that are directly related to the main cancer hallmarks. Among these functions are focal adhesion, cell cycle, PI3K-Akt signaling, insulin signaling, Ras and MAPK signaling pathways. Several hub genes and gatekeepers were found that could serve as potential drug targets for hepatocellular carcinoma. POU2F1 and PPARA showed a significant difference between low and high survival probabilities (P-value ⩽ .05) in HCC patients. Our study sheds light on important biomarker miRNAs for hepatocellular carcinoma along with their target genes and their regulated functions.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is one of the most common causes of cancer death worldwide. The incidence and mortality rates of HCC are increasing exponentially all over the world. 1 Generally, HCC occurs in the setting of underlying liver diseases such as hepatitis B and C as well as genetic and environmental factors.2-4 Hallmarks of cancer are represented by several main processes, including sustained proliferative signaling, bypassing growth suppressors, enabling replicative immortality, cell death resistance, angiogenesis, abnormal metabolic pathways, invasion/metastasis activation, and immune system evasion.5,6
Only about 20% to 30% of HCC patients undergo proper curative therapy. The lack of early reliable diagnostic markers and treatment strategies contributes to the high mortality rate of HCC highlighting the importance of reliable and accurate diagnostic markers. 7 Prompt diagnosis of HCC helps raise survival rates and avoid increased chemotherapy resistance and tumor recurrence. 8 Therefore, there is an urgent need to identify precise and effective biomarkers for prognosis and early diagnosis of HCC in addition to novel therapeutic tools.7,9
MicroRNAs (miRNAs) represent one of the most important and well-known classes of small non-coding RNA molecules. MicroRNAs are approximately 22 nucleotide long RNA molecules that act as guide molecules at the posttranscriptional level. 10 They are master regulators for protein-encoding genes that function by binding to their specific mRNA targets promoting either their degradation or translational inhibition. 10 miRNAs play major roles in a wide range of essential biological processes including cell proliferation, 11 cell differentiation, 12 apoptosis, 13 inflammation 14 and metabolic control. 15 Moreover, miRNAs have an effective role in tumorigenesis, acting as either tumor suppressors or oncomirs.16,17 It has been widely revealed that HCC initiation and progression are highly associated with dysregulated miRNA expression. 7 Therefore, studying differentially expressed miRNAs in HCC is a determinant step for exploring their role as biomarkers for early diagnosis or possible therapeutic tools for this debilitating disease.
Hence, miRNAs are among the most widely studied diagnostic and prognostic biomarkers of HCC. Researchers worldwide are trying to improve their understanding of miRNAs functionality in HCC. 18 One of the best ways that lead to a deep understanding of different miRNA roles is by performing a meta-analysis that facilitates summarizing and reviewing previous studies. Meta-analysis is a widely used technique that gives a comprehensive interpretation of a large amount of information and datasets which rise every day. 19
The present study aimed to discover significant biomarker miRNAs for HCC by performing computational analysis for a group of HCC miRNAs expression datasets of 147 tumor samples relative to 50 normal control samples, exploring their target genes, enriched pathways, and biological functions. As a result, we identified 34 significantly dysregulated miRNAs; 5 of which were down-regulated and 29 were up-regulated. The down-regulated miRNAs were miR-139-3p, miR-39-5p, miR-1258, miR-424-5p and miR-490-3p while the 5 most significantly up-regulated miRNA were miR-767-5p, miR-105-5p, miR-1269a and -b, miR-4652-5p, miR-183-5p, miR-182-5p, miR-96-5p and miR-10b-3p. Functional analysis for these miRNA targets presented several pathways and cellular processes that are directly related to main cancer/HCC hallmarks. Thus, dysregulation of these miRNAs may be implicated in liver cancer and may serve in miRNA-based diagnosis, prognosis, and therapy for such carcinoma.
Materials and Methods
Sequence reads collection
Liver sample data were obtained from The Cancer Genome Atlas (TCGA Research Network): https://www.cancer.gov/tcga. Samples were classified into 2 groups: (i) primary solid HCC tumors including 147 tumor samples, and (ii) normal tissues including 50 noncancerous samples. All accession numbers for tumor and normal samples were listed in Table S1. An overview of the present study workflow is shown in Figure 1.

The workflow of miRNA expression data analysis to unravel mechanisms underlying HCC progression. The blue rectangles represent methods that process input data (above) using computational tools (eg, YM500v2, DAVID) to produce the output (below).
Sequence reads processing and differential expression analysis
The YM500v220 algorithm was used to perform a meta-analysis for the 2 groups of samples (HCC and normal) using its user-friendly interface. YM500v2 algorithm is designed to connect to the TCGA data portal and retrieve sequences of interest to perform analysis. The YM500v2 tool also ensures the quality of the datasets. YM500v2 uses DESeq, R/Bioconductor package to analyze high-throughput sequencing data to get a differential expression. DESeq performs normalization and estimates the effective library size to obtain more accurate results. 21 All datasets were pre-processed to exclude the poor-quality reads. miRNA expression datasets meta-analysis was performed using the YM500v2 server to compare miRNA expression in normal and cancerous liver tissues. This data analysis was done on 147 tumor samples relative to 50 normal control samples obtained from TCGA and differentially expressed miRNAs with adjusted P-value ⩽ 0.05 were retrieved.
Target prediction
miRNAs target prediction was performed using miRWalk and miRror Suite in silico target prediction tools. mirWalk is a powerful comprehensive tool that provides a collection of both predicted and experimentally validated miRNA targets. Target prediction by miRWalk allows for identifying the common targets between different prediction algorithms. These algorithms use different criteria in predicting miRNA-target interaction such as seed base, thermodynamics, target accessibility, and folding energy.22,23 In this study, we selected the intersection between 5 algorithms within the miRWalk tool to perform our target prediction analysis including: mirWalk2.0, miRanda, PicTar2, RNA22v2, and Targetscan 6.2.
MiRror Suite database was used to obtain the combinatorial miRNA target genes based on the concept of combinatorial regulation by an ensemble of miRNAs. MeRtegrate protocol is the backbone of the MiRror Suite platform. 24 MiRror Suite provided a statistically ranked target list in a suitable format that was directly forwarded to the enrichment analysis tools.
Functional enrichment analysis
Enrichment analysis was performed using DAVID (The Database for Annotation, Visualization, and Integrated Discovery) functional enrichment tool.25,26 Our individually obtained gene sets (predicted and validated) by mirWalk as well as the combinatorially obtained ones by miRor were entered into the DAVID tool. Afterward, we obtained the enriched biological functions and KEGG pathways and screened for significantly enriched cancer/HCC-related terms.
Identification of TF- miRNAs regulatory interaction
To obtain the TF- miRNA regulatory relationship for each of our studied up-regulated and down-regulated miRNAs, we used the TransmiR v2.0 database, 27 through which we could detect the TFs that interact with our miRNAs.
Network construction and topological analysis
A network was constructed based on interactions among microRNAs, their targets, and transcription factors. We analyzed the network for topological properties using a Cytoscape plugin NetworkAnalyzer. 28 Topological properties provide useful information about network structure in general, and more specific node-associated properties (including node degree (ND) and betweenness centrality (BC)). ND is the number of edges (or links) connected to a node. ND allowed us to identify “hubs” in the network. Hubs are nodes with many more links than others. 29 BC of a node is the number of shortest paths from all nodes to all others that pass through that node. BC identifies gatekeeper nodes, which play a crucial role in the communication between different parts of a network.30,31
Molecular signatures and their validation through patient data
We performed patient data survival analysis of the highest ND (ie, hub) and BC (gatekeeper) nodes (from Tables 3 and 4) using the UCSC Xena browser (http://xena.ucsc.edu). 32 Samples were grouped based on the high and low expression of selected molecules to their median expression values. We used the default settings of the UCSC Xena browser to perform Kaplan-Meier survival analysis to find out which of the genes with the highest ND and BC nodes affects patient’s survival. The patients were stratified into low and high survival probability groups to determine if there is a statistically significant survival difference.
Results
Differential expression analysis
In the present study, the YM500v2 server 20 was used to get the significantly dysregulated miRNAs in normal and liver cancer tissues. Thirty-four significant deferentially regulated miRNAs in liver cancer compared to normal liver tissues were detected. The statistical results for the most significant miRNAs are shown in Table 1.
Significantly dysregulated miRNAs. The table shows 34 significantly dysregulated (up-regulated and down-regulated) miRNAs obtained from the YM500v2 server with adjusted P-value (P-adj ⩽ .05).

Target prediction analysis
Using the mirWalk tool we obtained the validated as well as predicted targets for each miRNA in our set with their P-values ⩽ .05 that resulted from the intersection of mirWalk2.0, miRanda, PicTar2, RNA22v2, and Targetscan 6.2. We then integrated all the targets together for undergoing the enrichment analysis. In addition, miRror Suite was used to perform a combinatorial target prediction analysis. We obtained a combination of common targets for our 34 input miRNAs (Table S2).
Enrichment analysis
Enrichment analysis for the predicted and validated targets obtained by mirWalk tool was done using the DAVID server. The results provided several significant cancer/HCC-related pathways and biological processes with Bonferroni corrected P-value ⩽ .05. Among the highly significant pathways are “pathways in cancer,” “PI3K-Akt signaling pathway,” “metabolic pathways,” “focal adhesion,” “MAPK signaling pathway,” “oxytocin signaling pathway” and “proteoglycans in cancer” with Bonferroni corrected p-value: 4.23E-09, 2.66E-07, 3.20E-07, 2.46E-05, 3.06E-05, 4.93E-05, and 5.15E-05 respectively (Figure 2A). Also, a number of highly significant biological processes were obtained such as transcription-related processes, signal transduction-related processes, response to drug, cell proliferation, and others with their Bonferroni corrected P-value ⩽ .05 (Table S3).

Bar charts illustrating the highly enriched pathways for the 34 dysregulated miRNA target genes: (A) illustration of the significantly enriched KEGG pathways of the validated and predicted target genes obtained by the mirWalk tool (Bonferroni corrected P-value ⩽ .05) and (B) illustration of the significantly enriched KEGG pathways of the combinatorially predicted target genes obtained by the mirRor tool (P-value ⩽ .05).
Predicted targets obtained from the miRror combinatorial prediction tool, were forwarded to the DAVID tool for performing the enrichment analysis. Our results revealed that combinatorial target genes of our miRNA set significantly regulate multiple cancer-related pathways with a P-value ⩽ .05 such as “melanoma,” “prostate cancer,” “PI3K-Akt signaling pathway,” “choline metabolism in cancer”, “glioma,” and others (Figure 2B). Moreover, our results revealed several cancer-related biological processes with a P-value ⩽ .05 the most significant of which are: “regulation of phosphatidylinositol 3-kinase signaling,” “axon guidance,” “ERBB2 signaling” and “positive regulation of cysteine-type endopeptidase activity involved in apoptotic process” (Table S4).
The enriched functions found to be common between collectively detected miRNA targets using the miRwalk tool and combinatorially detected ones using the miRror Suite tool are “PI3K-Akt signaling pathway,” “focal adhesion,” “insulin signaling pathway,” “Ras signaling pathway,” “Rap1 signaling pathway,” “cell cycle,” and “MAPK signaling pathway.” This indicates that dysregulated of the 34 miRNAs identified in our study induces the mentioned cancer/HCC-related processes.
Network construction and topological analysis
To elucidate the structure and functional roles of interacting components in a given cellular function (or malfunction), it is essential to initially organize data and knowledge about them in a network (ie, vertices connected to edges). We constructed a network from miRNA-gene and transcription factor-miRNAs interactions using the Cytoscape (Figure 3). The network contained 8992 nodes and22 091 interactions (File S1 is a Cytoscape.cys file of the network). Networks are the formalized representation of a large number of experimental studies in a diagrammatic format, which are computer-readable and can be analyzed using computational tools. We analyzed the network for topological properties using Cytoscape plugin Network Analyzer (Table 2). The networks containing few hubs are considered robust against single random perturbations (in Figure 3, nodes represented by large circles can be hubs that may act as potential drug targets). More specifically, Table 3 listed the top 10 highest degree nodes (TNRC6B, ZNF704, PPP1R12B, PRLR, QKI, RAB3B, LPP, PPARA, MECP2, PSD3, ADCY1). Table 4 shows the top 10 nodes with the highest BC which identifies gatekeeper nodes (ZNF704, PRLR, PPP1R12B, RAB3B, POU2F1, TNRC6B, PPARA, ST8SIA3, MECP2, and MTMR3).

A Network constructed based on miRNA-Gene and transcription factor-miRNA interactions using Cytoscape. The size of the nodes represents the value of the node degree. The node color ranges from green (low betweenness centrality) to red (high betweenness centrality).
Topological parameter values of the network.

The top 10 highest degree node (ND) genes and miRNAs.
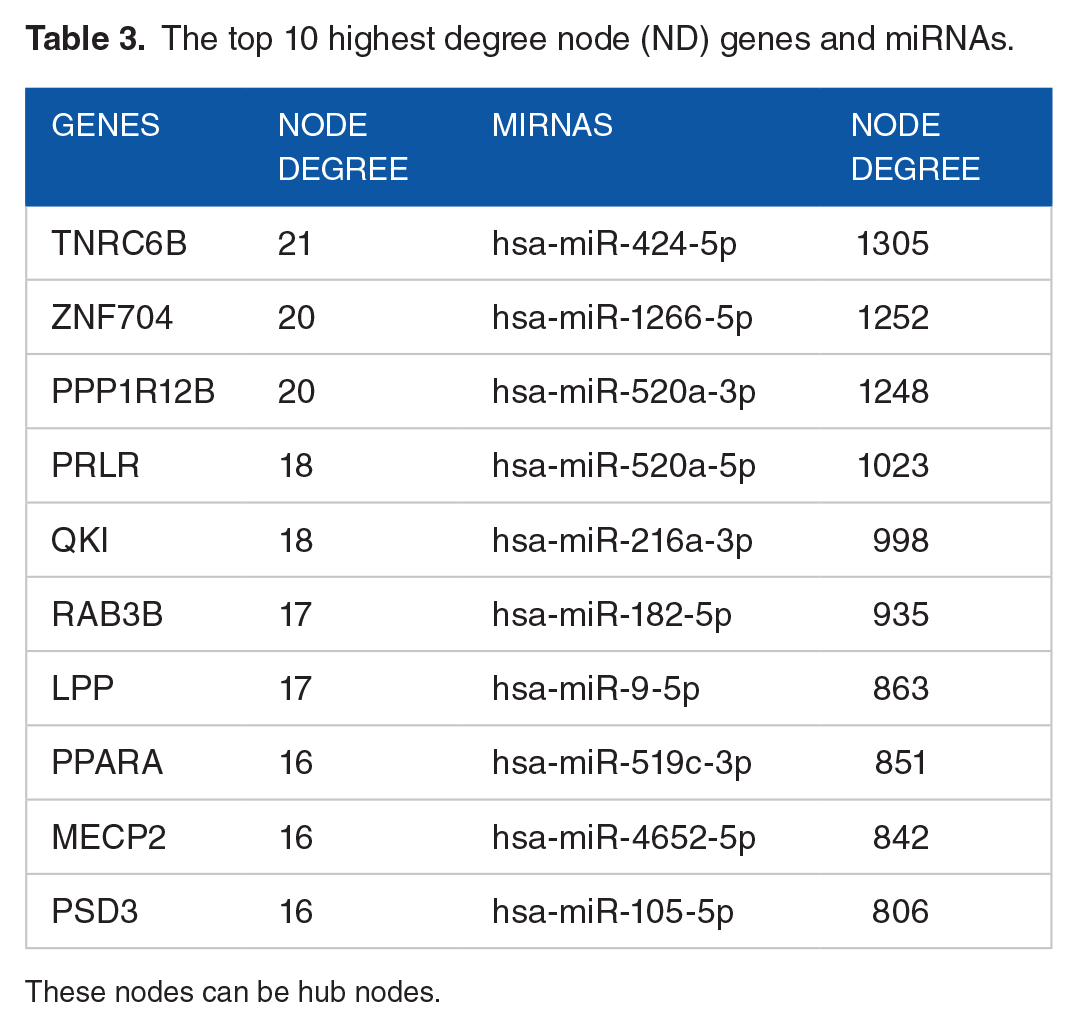
These nodes can be hub nodes.
The top 10 nodes (genes/TFs and miRNAs) with the highest betweenness centrality (BC).

Molecular signatures and their validation through patient data
The resulting top genes with the highest node degree (hubs) and those with the highest betweenness centrality (gatekeepers) shown in Tables 3 and 4, undergone survival analysis. Among these genes, POU2F1 and PPARA showed significant difference between low and high survival time in HCC patients (with P-value ⩽ .05). Patients with high expression of POU2F1 or low expression of PPARA exhibited low survival probability and vice versa (Figure 4). In our network PPARA is regulated by the following miRNAs: hsa-miR-424-5p, hsa-miR-1258, hsa-miR-9-5p, hsa-miR-96-5p, hsa-miR-135a-5p, hsa-miR-182-5p, hsa-miR-183-5p, hsa-miR-216b-5p, hsa-miR-519c-3p, hsa-miR-520a-3p, hsa-miR-520a-5p, hsa-miR-767-5p, hsa-miR-1266-5p, hsa-miR-1269a, and hsa-miR-1269b. While POU2F1 is regulated by hsa-miR-139-5p, hsa-miR-424-5p, hsa-miR-9-5p, hsa-miR-105-5p, hsa-miR-182-5p, hsa-miR-216a-5p, hsa-miR-224-5p, hsa-miR-452-5p, hsa-miR-519c-3p, hsa-miR-520a-3p, hsa-miR-1266-5p, hsa-miR-1269a, hsa-miR-1269b, and hsa-miR-4652-5p. These results suggest that POU2F1 and PPARA can be used as therapeutic targets for HCC.

Survival probability analysis of the highest ND and BC nodes. N is the number of samples in a group. _L represents low expression and _H represents high expression.
Discussion
This computational analysis was performed to compare miRNA expression in 147 liver cancer samples and 50 normal control samples. We reported 34 significantly (P-value ⩽ .05) dysregulated miRNAs for liver cancer; 5 were down-regulated and 29 were up-regulated. We then performed miRNA target prediction analysis followed by functional enrichment analysis of detected targets to find what pathway and biological process these miRNAs may affect. We found that most of the pathways were frequently associated with cell signaling and cancer pathogenesis. Our study provides in silico evidence on the implication of this dysregulated miRNA set in HCC onset and pathogenesis. These miRNAs may serve as diagnostic and therapeutic tools for such tumor.
Unlike our previous miRNA meta-analysis study, in which the studied miRNAs were collected from published miRNA profiling studies as well as the PhenomiR database in HCC versus non-HCC tissues, 16 miRNAs in the present study were obtained by YM500v2 server from TCGA data comparing miRNA expression data of HCC samples versus normal control samples.
Agreeing with some previous meta-analyses and miRNA profiling studies that screened miRNA expression in HCC our study defined a group of dysregulated miRNAs as possible characteristic biomarkers for HCC.16,26,33-45 A number of common miRNAs between the current study and previous comprehensive ones are shown in Table 5.
Hepatocellular Carcinoma (HCC) dysregulated miRNAs identified by the current study compared to previous meta-analysis and molecular studies.

Common miRNAs between the current study and previous ones are written in bold. The common downregulated miRNA was
The HCC down-regulated miRNAs identified in this study were miR-139-3p, miR-139-5p miR-1258, miR-424-5p, and miR-490-3p. miR-139 (miR-139-5p) was detected to be down-regulated in our analysis and multiple previous miRNA screening studies in HCC (Table 5). It was believed that for a certain microRNA hairpin structure (pre-microRNA) only one mature microRNA is produced (guide strand), while the other strand (passenger strand) is degraded. But now it has been revealed that sometimes the passenger strand does not degrade and both miR-5p/−3p strands may act as functional miRs and are implicated in the pathology of several types of cancer. 46 This is the case for miR-139-3p and miR-139-5p. In liver cancer, both were found to significantly suppress cell growth, migration, and invasion as well as being found to prevent metastasis of HCC cells. It was reported that miR-139-5p significantly reduced cell migration and invasion in part by downregulating the cell motility protein Rho-kinase 2 (ROCK2) in human HCC samples. 47 miR-139-3p was also found to reduce tumor growth and metastasis by targeting Annexin A2 Receptor (ANXA2R). miR-139-5p was found to inhibit epithelial-mesenchymal transition (EMT) in HCC by targeting ZEB1 and ZEB2; transcription factors that contribute to EMT. 48 It also prevents HCC cell growth by down-regulating karyopherin alpha-2 ( KPNA2). Moreover, miR-139-5p overexpression together with KPNA2 suppression decrease the level of the pro-oncogenes POU class 5 homeobox 1 (POU5F1) and -myc. 49 Also, both miR-139-3p and −5p have been shown to significantly inhibit cell migration and invasion in other types of cancer such as bladder cancer through targeting matrix metalloprotease 11 gene (MMP11) 50 and cervical cancer.51,52
Our result reported miR-1258 to be down-regulated in HCC. This downregulation was found to contribute to carcinogenesis and liver cancer progression through activation of its downstream target; CKS1B. Blocking the miR-1258-CKS1B axis was proposed as a possible therapeutic approach. 53 Our computational study also revealed the downregulation of miR-424-5p which was found to inhibit metastasis through attenuating cell resistance to anoikis, 54 a type of programed cell death that takes place in anchorage-dependent cells when they lose contact with their extracellular matrix or neighboring cells. 55 miR-424-5p was also found to prevent the EMT process and malignancy in HCC cells. miR-424-5p performs its anti-tumor actions via targeting the potent β-catenin inhibitor; ICAT and maintaining the E-cadherin/β-catenin complex. 56
Our analysis revealed that miR-490-3p is among the down-regulated miRNAs in the investigated test samples. It was demonstrated that miR-490-3p acts as a potential tumor inhibitor that suppress cell proliferation and enhance G1 cell cycle arrest and apoptosis in some cancers such as osteosarcoma, 57 ovarian cancer, 58 colorectal cancer 59 and breast cancer. 60 Agreeing with our results, previous studies reported a down-regulation for miR-490-3p in HCC,61,62 and that it suppresses HCC cell proliferation and migration by targeting the aurora kinase A gene (AURKA). 63 However, Zhang et al (2013) reported that miR-490-3p acts as an oncomir that is implicated in cell growth and epithelial to mesenchymal transition processes in HCC. 64 Hence, further deep studies concerning the role of mir-490-3p in cancer are strongly recommended.
On the other hand, the significant 29 up-regulated miRNAs revealed by our meta-analysis study involved a number of up-regulated miRNAs commonly identified with other meta-analyses and miRNA screening studies in HCC. They included miR-224, miR-216, miR-183/miR-182/miR-10b, miR-9, miR-96, miR-217/miR-1269b/miR-135a (arranged from the most common to the least common ones).
miR-224 is one of the most frequently reported HCC up-regulated miRNAs (oncomir). It enhances cell proliferation by targeting apoptosis inhibitor-5 (API-5) 26 and by activating AKT signaling pathway. 65 It also contributes to invasion and anti-apoptosis. 66 miR-224 was observed to be specifically up-regulated in the liver tissues of HCV-associated HCC, relative to healthy ones. 67 Lately, miR224 was considered an early diagnostic marker in plasma of patients with HCC preceded by chronic HCV infection. 68
miR-216a, also called miR-216 or miR-216a-5p according to mirbase-(http://www.mirbase.org/cgi-bin/mirna_entry.pl?acc=MI0000292) is another commonly detected HCC associated miRNA detected in various studies (Table 5). It was previously revealed that miR-216a-5p is up-regulated in multiple types of cancers, such as colorectal, prostate cancer, and others.69,70 It was demonstrated that miR-216a-5p contributes to cancer cell proliferation, viability, motility, and apoptosis in renal cell carcinoma. 70 In liver cancer, it was revealed that miRNA-216a is up-regulated by the androgen pathway suppressing the expression of TSLC1 in early liver carcinogenesis. 71 In addition, miR-216a/miR-217 cluster [both are located within the second intron of a non-coding RNA (RP23-298H6.1-001)], was previously identified in HCC. miR-216a/miR-217 cluster targets PTEN (phosphatase and tensin homolog) and SMAD7 leading to activation of the PI3K/Akt and TGF-β pathways which mediate EMT, cell migration, and stem-like properties of HCC cells, contributing to hepatocarcinogenesis and tumor progression. 72 In addition, miR-216a-5p was found to be involved in proliferation, viability, and cell motility in renal cancer. 70 On contrary, miR-216a-5p was shown to inhibit tumorigenesis in pancreatic cancer by targeting TPT1/mTORC1 and is mediated by LINC01133. Therefore, miR-216a-5p may have opposing actions in cancer according to the type of tumor 73 a matter that needs further studies in different types of cancer. The passenger strand of miR-216, miR-216a-3p was also found among the significantly up-regulated HCC miRNAs in our study which is a new finding that needs to undergo further investigation.
miR-217 is also detected to be up-regulated by our analysis, miR-217 was previously reported to promote cancer stem cell properties by targeting DKK1, activating the Wnt signaling pathway in HCC. 74 However, others suggested that miR-217 may function as a tumor suppressor in HCC by inhibiting the invasion of HCC cells via targeting E2F3 75 or targeting KLF. 76 Hence, deeper studies are required to explore the exact role of miR-217 in HCC.
Among the up-regulated HCC miRNAs identified by our study and found to be common with other previous studies are the miR-183 family members (miR-183, miR-96, and miR-182), however, mir-183 and miR182 are more frequently detected. This family represents a highly conserved miRNA cluster with sequence homology, whose members are located within a 5 -kb region on human chromosome 7q32.2.77,78 They are produced from a single primary transcript in the same direction from the telomere to the centromere. They possess similar biological functions that contribute to closely related signaling pathways. 79 miR-183-96-182 cluster was reported to have oncogenic effects in HCC and act as a prognostic tool. They promote migration and invasion by targeting FOXO1,79,80 while they promote cell proliferation via targeting miR-183 to PDCD4 and SOCS-6, targeting miR-96 to FOXO1 and FOXO3, and targeting miR-182 to CEBPA and TP53INP1. 79 However, this cluster was found to act as a tumor suppressor in other cancer types such as colon and lung cancers. 81
miR-10b is another commonly identified HCC up-regulated miRNA (Table 5). It enhances cell viability and invasion by targeting the tumor suppressor gene, CSMD1. 82 Moreover, miR-10b promotes cell proliferation, migration, and invasion by targeting HOXD10 and triggering the RhoC/ uPAR/ MMPs pathway. 83
miR-9 was also identified as HCC up-regulated miRNAs by several screening studies (Table 5). miR-9 is mainly a neurogenesis regulator, but its dysregulation was observed in cancer. 84 It was found to exert its oncogenic activities by regulating PPARA and CDH1 85 and to enhance HCC cell proliferation, migration, and invasion by targeting ESR1. 86 However, Han et al (2018) study suggested that miR-9 acts as a tumor suppressor in HCC. 87
Lastly, the current study shows up-regulated expression of miR-1269b and miR-135a in HCC agreeing with previous miRNA screening studies (Table 5). mir-1296b was indicated to contribute to carcinogenesis in HBV-associated HCC through promoting CDC40 (cell division cycle 40 homolog) expression, which in turn enhances cell growth, cell cycle progression, and cell migration in HCC cells. This takes place through the HBx/NF-κB/miR-1269b/CDC40 pathway, where HBx firstly stimulates NF-κB to enhance miR1269b expression. 88 miR-135a triggers cell migration and invasion in HCC by targeting FOXO1 (forkhead box O1) and enhancing the expression of MMP2 and Snail. 89 Recently, miR-135a upregulation was noticed to be associated with HCC recurrence which renders it a novel biomarker for patients needing adjuvant therapy after resection. 90
Interestingly, our study revealed highly significant dysregulated miRNAs including miR-767-5p, miR-105-5p, miR-1269a, and miR-4652-5p, which may need more focused investigations regarding their roles in HCC. miR-767-5p was found to be among a group of migration-facilitating miRNAs. 91 Moreover, miR-767 was reported to target the tumor suppressor genes; TET1 and TET3, hence playing a tumor-promoting role in cancer. 92 Recently, miR-767-5p was reported as a potent oncomir in HCC that acts in part by repressing PMP22 signaling. 93
miR-105 (also known as miR-105-5p) was considered to have a cancer-specific effect where it enhances cancer cell survival and metastasis. Cancer-secreted miR-105 can breakdown tight junctions of vascular endothelial barriers triggering metastasis. 94 An oncogenic effect for miR-105-5p was detected in many cancer types such as gastric cancer, lung cancer, and colorectal cancer. 95 Conversely, miR-105 has been reported as a tumor suppressor in HCC. 96 Therefore, further investigations are required to clarify the role of miR-105 in HCC.
We also identified miR-1269a and its sister miR-1269b, as significantly HCC up-regulated miRNAs. miRNA sisters that differ by only 1 or 2 nucleotides receive different alphabetical suffixes. 97 They were recently identified as emerging oncomirs that are upregulated in HCC. 98 miR-1269a was found to target CXCL9, SOX6, VASH1, ATRX, RASSF9, SMAD7, HOXD10, and FOXO1, while miR-1269b targets METTL3, CDC40, SVEP1, and PTEN. 98 It was determined that miR-1269 directly acts on FOXO1 promoting proliferation and tumorigenicity of HCC cells. 99 Importantly, both miR-1269a and miR-1269b represent excellent diagnostic and prognostic markers for HCC. 98
miR-4652-5p was determined as a novel significantly up-regulated miRNA in HCC by our analysis. This miRNA was recently detected among cancer-inducing miRNAs in head and neck squamous cell carcinoma, 100 however, it needs further experimental validation to investigate its role.
For miRNA target analysis, we used 2 miRNA target analysis tools for the differentially expressed miRNAs as previously described 101 : miRror, which is a combinatorial miRNA target prediction tool, and miRWalk, which provides the predicted and experimentally validated targets for each miRNA. Within miRWalk, we used the intersection of different prediction algorithms (mirWalk2.0, miRanda, PicTar2, RNA22v2, and Targetscan 6.2) to obtain the most probable targets and minimize false positives. Then we integrated the resulting target genes of miRWalk. The main purpose for using both tools was to have an integrative view of combinatorial and individual microRNAs interactions and hence functions and pathways.
Functional enrichment analysis for the miRNA targets obtained by both the collective and the combinatorial methods provided a number of significant HCC-related pathways and biological processes. The obtained enriched functions involving “PI3K-Akt signaling pathway,” “focal adhesion,” “insulin signaling pathway,” “Ras signaling pathway,” “Rap1 signaling pathway,” “cell cycle,” and “MAPK signaling pathway” were commonly detected by the collective and the combinatorial target analysis methods. PI3K-Akt signaling pathway or phosphatidylinositol 3′–kinase (PI3K)-Akt signaling pathway regulates important cellular processes such as transcription, translation, proliferation, growth, and survival. The binding of growth factors to their receptor tyrosine kinase (RTK) or G protein-coupled receptors (GPCR) stimulates type Ia and Ib PI3K isoforms, respectively. PI3K catalyzes the production of phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3, in turn, serves as a second messenger that helps to spark off Akt. Once active, Akt can manipulate key signals phosphorylating substrates implicated in apoptosis, protein synthesis, metabolism, and cellular cycle. 102 Importantly, the PI3K/Akt pathway, which is a prototypic survival pathway, greatly contributes to hepatocarcinogenesis. 103 It was reported that the activation of the PI3K/Akt signaling pathway causes EMT and multi-drug resistance in HCC cells.104,105
Focal adhesion or cell-matrix adhesion is a structural linking between the actin cytoskeleton and extracellular matrix where there are cell-extracellular matrix contact points, (focal adhesions) that form bundles of actin filaments anchored to transmembrane receptors of the integrin household. 106 It plays crucial roles in vital cellular processes including cellular adhesion, cell motility, intracellular signaling pathways, proliferation, differentiation, regulation of gene expression and survival. Focal adhesion is composed of various protein components such as receptors, structural proteins, adaptors, GTPase, kinases, and phosphatases.106,107 In HCC, the focal adhesion signaling pathway is implicated in tumor cell motility, metastasis, invasion, and survival as well as evasion of the anti-tumor immune response. 108 Another cell adhesion pathway detected by our enrichment analysis is Rap1 signaling. Ras-associated protein-1 (Rap1) is a small GTPase that regulates basic cellular functions such as cell-cell adhesions and junctions, migration, and polarization. Rap1 cycles between binding to GDP (inactive) and GTP (active) conformation. This process is controlled by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs).109,110 In cancer cells, Rap1 signaling is the main effector process in cancer cell invasion, and metastasis. 111
Insulin is primarily a hormone that is responsible for glucose uptake and controlling its level in blood. The insulin signaling pathway starts with insulin binding to its receptor (INSR) on the surface of target cells. The insulin signaling pathway mediates both the metabolic and mitogenic effects of insulin. 112 However, it has been observed that abnormal insulin levels and insulin-mediated signaling can lead to cancer development and progression. Insulin was found to exert oncogenic effects on the proliferative and anti-apoptotic pathways in cancer. In HCC cells, INSR is usually overexpressed and insulin signaling is known to be enhanced.112,113
Ras pathway represents a vital signaling network that regulates cell proliferation, growth, and survival. 102 RAS signaling is activated by binding of different growth factors to their receptors leading to the activation of c-raf, MEK, and ERK. In cancer, abnormal activation of RAS takes place due to several molecular alterations, such as methylation of tumor suppressors and amplification of oncogenes. Ras signaling is one of the therapeutic target pathways in HCC that when blocked, improvement of patient survival can be achieved. 114 MAPK (mitogen-activated protein kinase) signaling pathway involves different kinase proteins that link extracellular signals to the intracellular machinery. It mediates multiple cellular processes such as proliferation, growth, differentiation, apoptosis, and migration.102,115,116 MAPK cascade is known to play a critical role in the incidence, development, and progression of cancer. Regarding HCC, it was reported that the MAPK cascade activation contributes to drug resistance exerted by liver cancer cells. 117 Collectively, it has been shown that dysregulation of the Ras/MAPK signaling pathways, is strongly implicated in malignant transformation and cancer pathogenesis in the liver, where they are activated in 50-100% of HCC cells, and consequently, Ras/MAPK pathway effectors can act as potential targets for HCC therapy. 118
Further, we constructed a network from miRNA-gene and transcription factor-miRNAs interactions and performed a topological analysis to identify hub nodes and gatekeeper nodes using the Cytoscape plugin. To unravel the role of highest node degree (ND) and betweenness centrality (BC) nodes in HCC, we performed Kaplan-Meier survival analysis using TCGA patient data. Among them, POU2F1 and PPARA successfully stratified patients between low and high survival probability. Patients with high expression of POU2F1 or low expression of PPARA have low survival probability and vice versa.
POU2F1 (POU domain, class 2, transcription factor 1), also known as octamer-binding transcription factor 1 (OCT-1), is implicated in regulating the physiological and pathological processes of cancer cell as well as inflammatory processes and maintenance of tumor stem cells. 119 PPARA (peroxisome proliferator-activated receptor alpha) is a ligand-activated transcription factor that is highly expressed in liver under normal condition and is activated by fatty acids or any other lipid species. 120 POU2F1 overexpression was shown to enhance cell growth and EMT and to correlate to poor prognosis in HCC 121 while low expression of PPARA induces unfavorable outcomes in such malignancy. 122 Our results suggest that miRNAs regulating POU2F1 (including miR-139-5p, miR-424-5p, miR-9-5p, miR-105-5p, miR-182-5p, miR-216a-5p, miR-224-5p, miR-452-5p, miR-519c-3p, miR-520a-3p, miR-1266-5p, miR-1269a, miR-1269b, and miR-4652-5p) and PPARA (including miR-424-5p, miR-1258, miR-9-5p, miR-96-5p, miR-135a-5p, miR-182-5p, miR-183-5p, miR-216b-5p, miR-519c-3p, miR-520a-3p, miR-520a-5p, miR-767-5p, miR-1266-5p, miR-1269a, and miR-1269b) requires further attention to be used as potential HCC drug targets.
Conclusion
Studying miRNA expression in HCC may help to identify new biomarkers that may help in prognosis, early diagnosis, and discovering novel therapeutic targets for HCC. Our computational analysis presents a set of 34 significantly dysregulated miRNAs in liver cancer. Functional enrichment analysis of their targets has shown the implication of these 34 miRNAs in a variety of biological processes and pathways that are directly linked to cancer hallmarks. This study helps explore diagnostic and prognostic biomarkers for HCC and provides insights into miRNA-based HCC therapy. The study suggests also that POU2F1 and PPARA can be used as potential HCC drug targets.
Supplemental Material
sj-cys-5-cix-10.1177_11769351231171743 – Supplemental material for In Silico Analysis of MicroRNA Expression Data in Liver Cancer
Supplemental material, sj-cys-5-cix-10.1177_11769351231171743 for In Silico Analysis of MicroRNA Expression Data in Liver Cancer by Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M. Khan and Mahmoud Elhefnawi in Cancer Informatics
Supplemental Material
sj-docx-1-cix-10.1177_11769351231171743 – Supplemental material for In Silico Analysis of MicroRNA Expression Data in Liver Cancer
Supplemental material, sj-docx-1-cix-10.1177_11769351231171743 for In Silico Analysis of MicroRNA Expression Data in Liver Cancer by Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M. Khan and Mahmoud Elhefnawi in Cancer Informatics
Supplemental Material
sj-docx-2-cix-10.1177_11769351231171743 – Supplemental material for In Silico Analysis of MicroRNA Expression Data in Liver Cancer
Supplemental material, sj-docx-2-cix-10.1177_11769351231171743 for In Silico Analysis of MicroRNA Expression Data in Liver Cancer by Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M. Khan and Mahmoud Elhefnawi in Cancer Informatics
Supplemental Material
sj-docx-3-cix-10.1177_11769351231171743 – Supplemental material for In Silico Analysis of MicroRNA Expression Data in Liver Cancer
Supplemental material, sj-docx-3-cix-10.1177_11769351231171743 for In Silico Analysis of MicroRNA Expression Data in Liver Cancer by Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M. Khan and Mahmoud Elhefnawi in Cancer Informatics
Supplemental Material
sj-docx-4-cix-10.1177_11769351231171743 – Supplemental material for In Silico Analysis of MicroRNA Expression Data in Liver Cancer
Supplemental material, sj-docx-4-cix-10.1177_11769351231171743 for In Silico Analysis of MicroRNA Expression Data in Liver Cancer by Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M. Khan and Mahmoud Elhefnawi in Cancer Informatics
Footnotes
Acknowledgements
The HCC tissue data shown here are generated by the TCGA Research Network.
Funding:
The author(s) acknowledge funding from Academy of Scientific Research and Technology, Ministry of Scientific Research and Technology grant number #5202 PRISM.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
ME conceptualized the study and the methodology. NA and EH performed the analysis and wrote the manuscript. FK performed part of the analysis and wrote part of the manuscript. ME supervised the whole study. ME and NA performed the final revesion and editing of the paper.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.