
Abstract
BACKGROUND AND OBJECTIVE:
Osteosarcoma is the most common primary malignancy in bone. Patients who respond poorly to induction chemotherapy are at higher risk of adverse prognosis. The molecular basis for such poor prognosis remains unclear. Recently, increasing evidence has suggested decreased expression of miR-34a is observed in a number of cancer types, including human osteosarcoma, and decreased miR-34a is involved in drug resistance. However, the underlying molecular mechanisms of decreased miR-34a on cisplatin chemoresistance in osteosarcoma has not been reported.
METHODS:
Osteosarcoma U2OS cells were transfected with miR-34a mimics for 48 h, then the cells were treated with 3.0
RESULTS:
Treatment of U2OS cells with cisplatin induced cell apoptosis by upregulation of c-Myc -dependent Bim expression; Osteosarcoma U2OS cells transfected with miR-34a mimics (miR-34a/U2OS) induced cell apoptosis and inhibited cell survival, and increased the sensitivity of U2OS cells to cisplatin. U2OS cells transfected with miR-34a mimics upregulated the protein expression of c-Myc and Bim. Targeting c-Myc downregulated the expression of Bim in the miR-34a/U2OS cells. In addition, Targeting Bim reversed the chemeresistance of miR-34a/U2OS cells to cisplatin.
CONCLUSIONS:
Our data indicated that miR-34a enhanced the sensitivity to cisplatin by upregulation of c-Myc and Bim pathway.
Introduction
Osteosarcoma (OS) remains the most common primary malignant bone cancer affecting children and adolescents [1]. The 5-year survival for patients with localized OS remains at 60–70% with multiagent chemotherapy treatment together with surgical techniques [2]. Up to 20–25% of patients present with intrinsic or acquired resistant to chemotherapy, leading to recurrence and metastasis. However, the underlying mechanisms of chemotherapy resistance are still unknown [3].
MicroRNAs (miRNAs, miRs) are a family of small noncoding RNAs that regulate gene expression by sequence-selective targeting of mRNAs, leading to a translational repression or mRNA degradation [4]. Experimental evidence demonstrates that dysregulation of specific miRNAs leads to drug resistance in different cancers and correction of these miRNAs using miRNA mimics or antagomiRs can normalize the gene regulatory network and signaling pathways and sensitize cancerous cells to chemotherapy [5, 6, 7, 8].
miR-34a is a member of an evolutionarily conserved miRNA family, miR-34s. It has been shown to repress several oncogenes directly or indirectly [9, 10]. Ectopic miR-34a expression resulted in cell cycle arrest and growth inhibition and attenuated chemoresistance to anticancer drug camptothecin by inducing apoptosis, suggesting a potential role of miR-34a for the treatment of prostate cancer [11]. Functional analysis of miR-34a in EWS cell lines indicated that restoration of miR-34a activity may be useful to decrease malignancy and increase tumour sensitivity to doxorubicin and vincristine, so sparing excessive long-term toxicity to EWS patients [12]. Zhao et al. [13] has recently reported that combination of doxorubicin chemotherapy and miR-34a replacement therapy synergistic antiproliferative effects and it is more effective than monotherapy in suppressing Ewing’s sarcoma xenograft tumor growth. However, the mechanisms of miR-34a sensitizes osteosarcoma cells to chemotherapy have not been totally elucidated. Therefore, it is of great significance to further study the function and mechanism of miR-34a in osteosarcoma.
c-Myc is a transcriptional activator implicated in the control of cell proliferation, differentiation and transformation, but is also involved in the regulation of programmed cell death, apoptosis [14]. Ectopic overexpression of c-Myc in both Rat1 fibroblasts and human osteosarcoma cells causes a dramatic increase of cell apoptosis [15]. Cerquetti et al. [16] has reported that silencing of c-Myc mRNA prevented paclitaxel induced apoptosis in SW13 cells, whereas in the H295R cells the overexpression of c-Myc rendered the cells more prone to growth inhibition after paclitaxel exposure.
A growing body of evidence supports a central role for the BCL2 homologous (BH) family of proteins in mediating MYC induced apoptosis. This family of proteins is subdivided into anti-apoptotic, pro-apoptotic BH3-only, and effector BH3 proteins, BAX and BAK. In response to pro-apoptotic stimuli, BAX and BAK oligomerize to form pores in the outer mitochondrial membrane (MOMP), triggering release of Cytochrome C, SMAC/DIABLO and consequent activation of effector Caspases [17, 18].
The recent identification of Bim as a transcriptional target of MYC suggests that this BH3-only protein may directly mediate MYC’s pro-apoptotic signal [19]. MYC induces Bim accumulation in Burkitt’s Lymphoma and MYC point mutants that fail to induce Bim also fail to induce apoptosis [20]. Muthalagu et al. has reported that
MYC induces apoptosis in postmitotic cells by upregulation of BIM [21]. Delbridge et al. has reported that MYC accelerated lymphomagenesis by downregulation of BIM [22]. In breast cancer cells, reduction of c-Myc expression by inhibition of mTORC1 activity abrogates occupancy of the Bim promoter by c-Myc, decreases Bim expression and promotes tolerance to Mcl-1 depletion [23]. Jiang et al. has reported that uberoylanilide hydroxamic acid (SAHA) induced apoptosis in rat fibroblast cells by selectively inducing the expression of Bim, leading to Bax activation in c-Myc-expressing cells. Where c-Myc was absent, Bim, despite its induction by SAHA, failed to activate Bax and was unable to induce apoptosis. These results indicate that c-Myc is dispensable for Bim induction by SAHA [24]. Egle et al. has reported that Bim is induced by Myc in B cells and mediates apoptosis. Inactivation of even a single allele of Bim accelerated Myc-induced development of tumors, particularly acute B cell leukemia [25]. Therefore, it is necessary, and maybe sufficient, to therapeutically impact on the Mcl-1/Bim balance for efficient induction of cancer cell death.
It has previously found that miR-34a functions as a potent tumor suppressor through the modulation of c-Myc in cellular senescence in HCC cells [26]. Yamamura et al. [27] reported that miR-34a was downregulated in prostate cancer tissues and silenced the expression of the c-Myc oncogene by targeting its 3’ UTR, inhibiting cell proliferation, cell invasion and promoting apoptosis. However, whether miR-34a induces apoptosis by c-myc/Bim signals remain still elusive.
In the present study, the antitumor effects of miR-34a were analyzed in osteosarcoma cells in vitro. The results show that overexpression of miR-34a induced osteosarcoma cells apoptosis and sensitized osteosarcoma cells to cisplatin by upregulation of c-Myc/Bim signal. These observations provide a promising gene therapeutic agent that miR-34a functions as a tumor suppressor gene, which could increase the susceptibility of osteosarcoma cell lines to cisplatin through up-regulating c-Myc/Bim signal in osteosarcoma.
Materials and methods
Cell culture
Human osteosarcoma U2OS cell line was purchased from the American Type Culture Collection (ATCC, Shanghai, China). The cells were cultured in F-12K or DMEM (Gibco, NY, USA) supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO
miR-34 a transfection
U2OS cells were plated a day prior to transfection. When cells reached 80% confluency, cultures were transfected overnight with miR-34a mimics and negative controls (scrambled oligos) (Life Technologies) at a final concentration of 100 nmol/l using Lipofectamine 2000. The transfection media were replaced with fresh media and cells harvested 48 h after transfection for functional analysis. To acquire the stable clones, 48 h after transduction, U2OS cells were selected with 400
siRNA transfection
U2OS cells, miR-34a overexpressing U2OS cells and scrambled oligos overexpressing U2OS cells were seeded on 60 mm dishes at 2
Quantitative RT PCR analysis of cell RNA
RNA was isolated from cells using TRIzol reagent and DNA prepared using TaqMan MicroRNA Reverse Transcription Kit (Invitrogen) according to the manufacturer’s protocol. Expression of miR-34a was assessed by quantitative reverse transcription PCR (RT-qPCR), using
Western blot analysis
Cells were lysed using radioimmunoprecipitation (RIPA) assay lysis buffer (PBS containing 1% NP40, 0.5% Na-deoxycholate, and 0.1% SDS) supplemented with 1
MTT assay
U2-OS cells (2
AnnexinV fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining assay
Flow cytometry was used to discriminate between intact and apoptotic cells. U2-OS cells were stained with fluorescein isothiocyanate (FITC) labeled annexinV that binds to membrane phosphatidylserine and with propidium iodide (PI) that binds to cellular DNA according to the manufacturer’s instructions (BD bioscience, USA). Briefly, the U2-OS cells (following siRNA transfection or/and miR-34a transfection for 48 h and treated with 3.0
Statistical analysis
All data were expressed as mean
Results
Cisplatin treatment upregulats c-Myc-dependent Bim expression
Osteosarcoma U2OS cells were treated with 30
Activation of c-Myc/Bim by cisplatin confers cisplatin sensitivity in U2OS cells. A, Western blot analyses of c-Myc and Bim expression in U2OS cells treated with 3.0 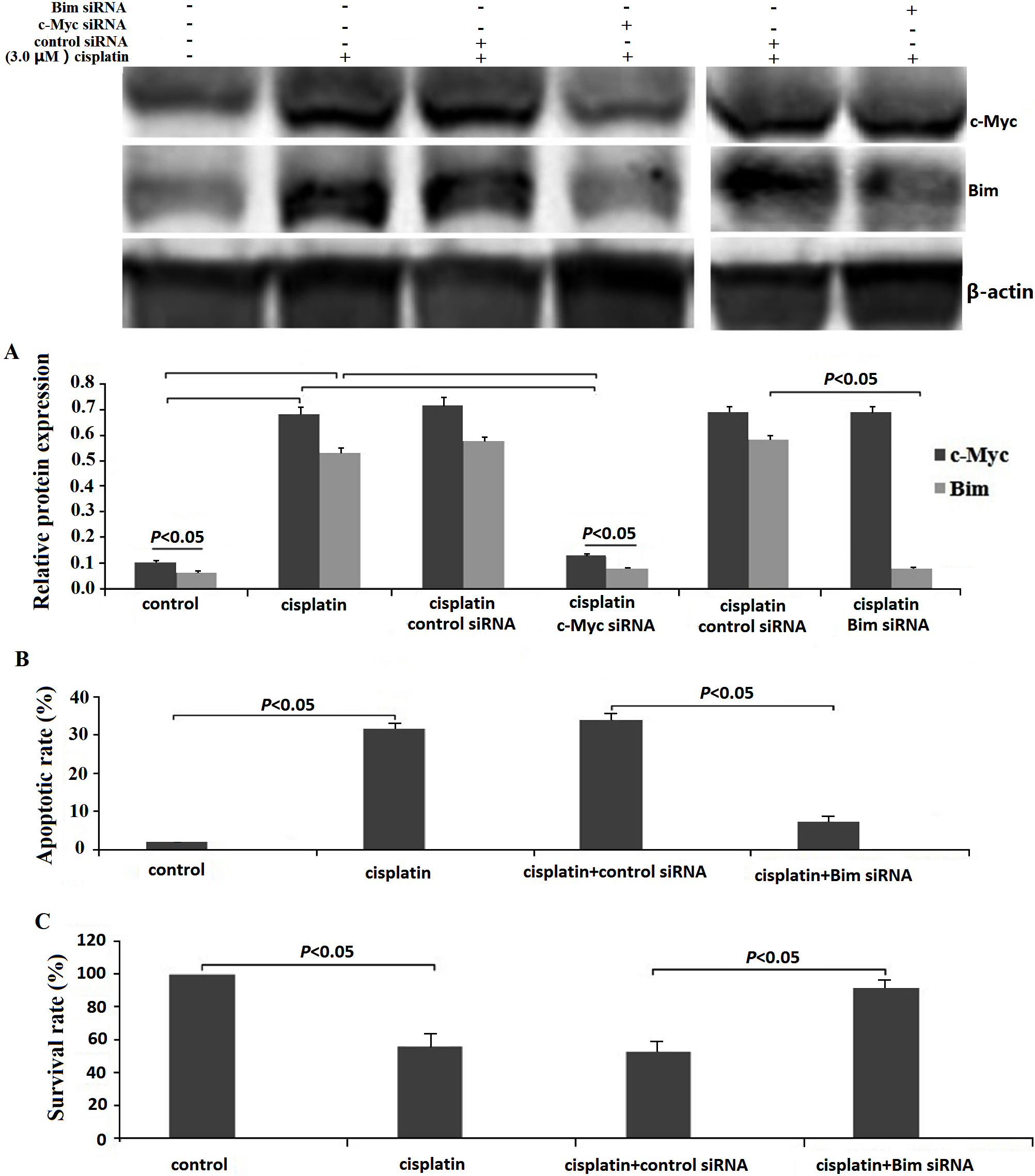
U2OS cells were treated with 3.0
c-Myc/Bim activation is essential for miR34a-induced apoptosis
To investigate the function of miR-34a in U2OS cells tumorigenesis, we transfected miR-34a mimics or miR-Ctrl in U2OS cells and determined their miR-34a levels 48 h after transfection. Results showed increased miR-34a levels in miR-34a transfected U2OS cell compared to miR-Ctrl transfected cells (Fig. 2A).
The role of miR-34a in the regulation of proliferation and apoptosis in U2-OS cells. A, U2OS cells were transfected with miR-34a mimics or miR-Ctrl for 48 h. miR-34a level was determined by qRT-PCR assay. B, Western blot analyses of c-Myc and Bim expression in U2OS cells transfected with miR-34a in the presence or absence of c-Myc siRNA transfection for 48 h, 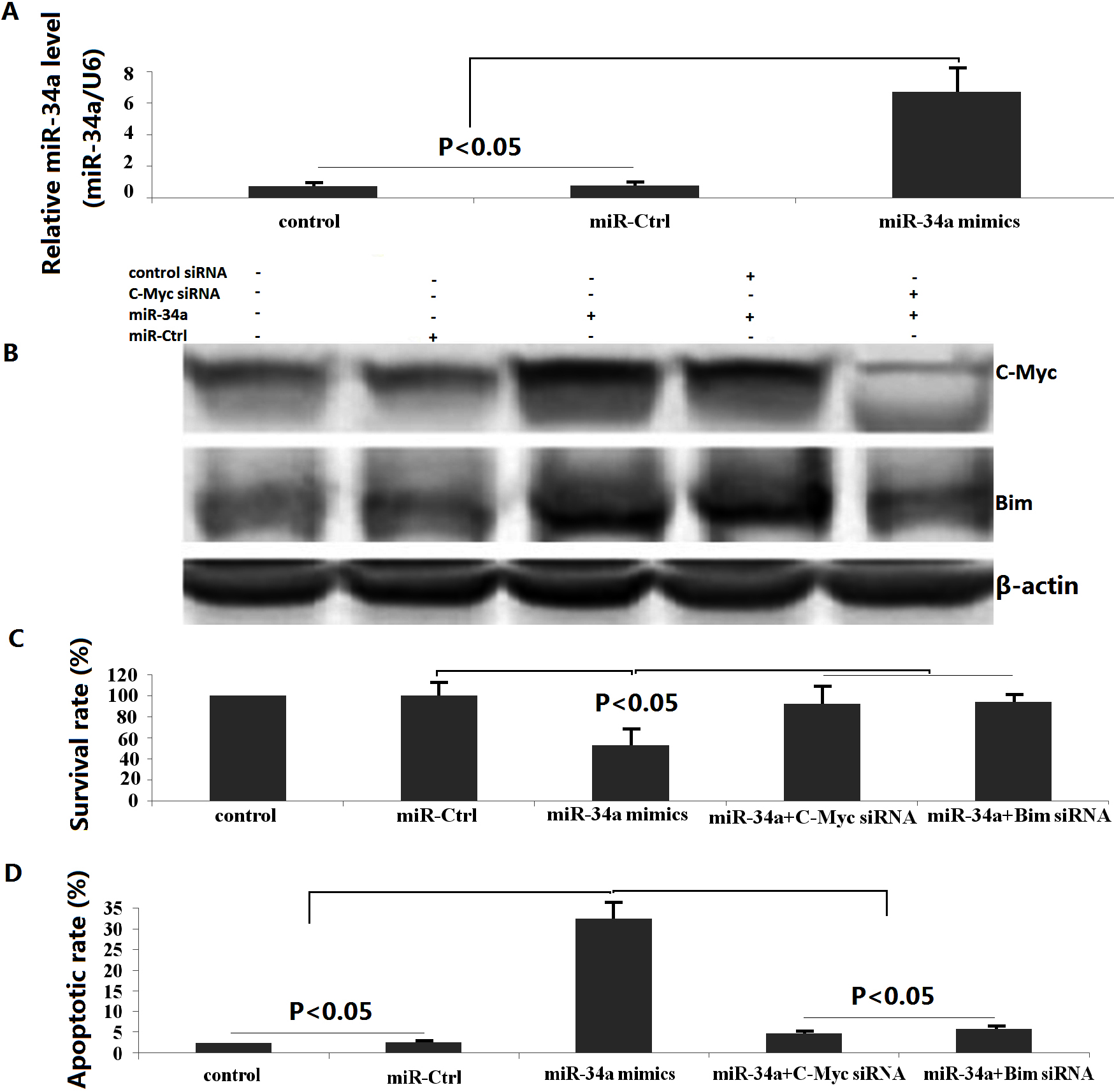
c-Myc and Bim protein levels were analyzed by western blot to investigate the mechanism by which miR-34a induced apoptosis in U2OS cells. c-Myc and Bim protein were significantly increased in U2OS cells, which express lower levels of endogenous c-Myc and Bim after transfection with miR-34a mimics (Fig. 2B).
We then investigated the effect of miR-34a on U2OS cell viability. As shown in Fig. 2C, miR-34a significantly inhibited U2OS proliferation by MTT assay. To investigate whether miR-34a affects U2OS cell apoptosis, U2OS cells were transfected with miR-34a and apoptosis assessed 24 h later. As shown in Fig. 2D, miR-34a transfected U2OS cells had a higher incidence of apoptosis than miR-Ctrl transfected U2OS cells by Flow cytometric analysis (FCM). These results suggest that miR-34a inhibits U2OS cell proliferation and induces apoptosis.
To further verify that the effects of miR-34a on proliferation and apoptosis in U2OS cells were mediated by regulation of c-Myc/Bim signal, we silenced Bim or c-Myc in U2OS cells by using siRNA, respectively. As shown in Fig. 2, knockdown of Bim reversed miR-34a-induced U2OS cell proliferation (Fig. 2C) and inhibited cell apoptosis (Fig. 2C), which was consistent with the effects of targeting c-Myc in miR-34a transfected U2OS cells (Fig. 2C–D).
To test whether c-Myc is involved in activating Bim during miR-34a-induced apoptosis, an siRNA strategy was utilized to knock down the expression of c-Myc in human osteosarcoma U2-OS cells. After 48 h of transient c-Myc siRNA transfection, c-Myc and Bim levels were reduced 80–90% (Fig. 2B). These data indicate that miR-34a upregulated c-Myc/Bim signal under these experimental conditions. We also showed that cisplatin treatment did not induced miR-34a expression in U2OS cells, indicating that miR-34a is not involved in cisplatin-induced apoptosis (data not shown).
miR-34a sensitizes U2-OS cells to cisplatin by activation of c-Myc/Bim pathway. A, MTT analyses of cell growth inhibition after miR-34a transfection and cisplatin treatment in the presence or absence of Bim siRNA or c-Myc siRNA transfection for 48 h. B, Flow cytometric analysis of cell apoptosis after miR-34a transfection and cisplatin treatment in the presence or absence of Bim siRNA or c-Myc siRNA transfection for 48 h. C, Western blot analyses of c-Myc and Bim expression in U2OS cells. 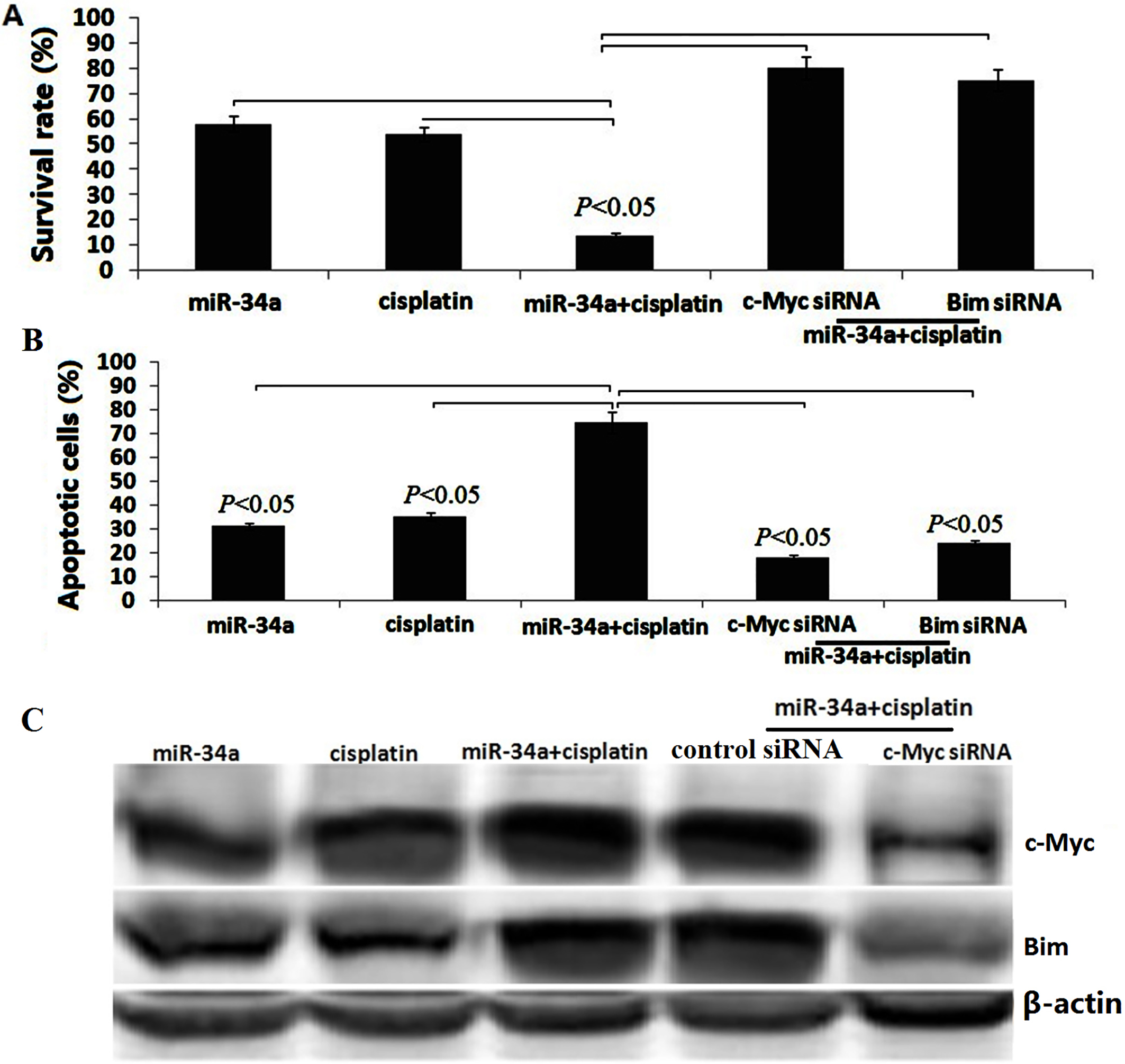
We next determined the effects of miR-34a overexpression on cisplatin sensitivity in U2OS cells. Briefly, U2OS cells were transfected with a miR-34a mimic or a miR-Ctrl for 48 h, then exposed to 3.0
miR-34a sensitizes U2-OS cells to cisplatin by activation of c-Myc/Bim pathway
miR-34a or a miR-Ctrl transfected U2OS cells were transfected with c-Myc siRNA or Bim siRNA or control siRNA for 48 h, then exposed to 3.0
In order to determine whether the observed enhancement in sensitivity to cisplatin following overexpression of miR-34a was due to its induction of c-Myc-dependent Bim, the miR-34a or a miR-Ctrl transfected U2OS cells were transfected with Bim siRNA or control siRNA for 48 h, then exposed to 3.0
Discussion
miR-34a was reported as a tumor suppressor. Furthermore, miR-34a was found to inhibit cancer stem cells (CSCs) self-renewal [28] and invasion [29] promoting their sensitivity to chemo- and radioth-erapy [28], providing evidence that miR-34a may function as an anti-oncogene. In the present study, we found that U2OS cells over-expressing miR-34a inhibited cell proliferation, decreased colony formation rate and induced apoptosis. The findings indicated that identification of miR-34a as a potent tumor suppressor is a highly significant finding with respect to the development of potential therapeutics for cancer. Although it is well accepted that miR-34a is a tumor-suppressor gene, the miR-34a KO mice are not tumor prone [30]. The tumor-suppressive function of miR-34a might be restricted to specific tissues and loss of miR-34a might cooperate with specific oncogenic lesions.
Cisplatin, which is widely used in chemotherapeutic therapy, is effective for treating patients with osteosarcoma [31], markedly increasing the survival rate. However, via various mechanisms, chemoresistance is one of the main obstacles preventing this increase [32]. It is well known that miRNA dysregulation influences tumor malignant progression. It has been demonstrated that miR-34a also play important roles in osteosarcoma chemoresistance [33, 34]. Consequently, our purpose was to elucidate the mechanism of cisplatin resistance and to discover possible means of improving osteosarcoma treatment. We demonstrated in our study that miR-34a overexpression enhanced the sensitivity of U2OS cells to cisplatin in vitro. Additionally, we showed that miR-34a overexpression enhanced cisplatin induced apoptosis in U2OS cells. These results suggest that miR-34a overexpressing therapies could be effective for the treatment of cisplatin-resistant osteosarcoma.
The c-Myc transcription factor, which regulates 15% of all human genes, plays an important role in a myriad of biological processes including cell growth and proliferation, cell cycle progression and apoptosis [35, 36, 37]. In other tumor types, c-Myc expression levels have been associated with drug resistance [38, 39, 40]. However, in other tumor types, c-Myc expression levels have been associated with drug sensitivity [41, 42, 43]. We demonstrated in our study that miR-34a overexpression upregulated c-Myc expression in U2OS cells, and accelerated cisplatin-induced apoptosis. However, targeting c-Myc decreased miR-34a induced apoptosis in U2OS cells, indicating that c-Myc is required for the synthetic induction of apoptosis driven by the combination of miR-34a and cisplatin.
Proapoptotic BH3-only proteins such as Bim bind to antiapoptotic proteins and thus allow the proapoptotic multidomain proteins, Bax and Bak, to form channels on the mitochondrial membrane leading to cytochrome
In ovarian cancer, degradation of Bim plays an important role in cisplatin resistance, and treatment of ovarian cancer cells with cisplatin caused Bim phosphorylation and subsequent degradation and that its degradation is associated with cisplatin resistance [46]. We demonstrated in our study that treatment of U2OS cells with cisplatin did not activate miR-34a, but caused the activation of c-Myc and Bim. By inhibiting c-Myc expression with siRNA, we show that Bim expression were blocked, which suggests that Bim is positively regulated by c-Myc. It has been shown that Bim transcriptional repression may mediate tumor chemoresistance [47]. Our data reveal that treatment of U2OS cells with cisplatin induced apoptosis that was suppressible by siRNA to Bim, suggesting that Bax is the primary mediator of cisplatin -induced apoptosis in U2OS cells.
In conclusion, the results show that miR-34a suppressed osteosarcoma cells growth and induced apoptosis by up-regulation of c-Myc-dependent Bim expression. Treatment of U2OS cells with cisplatin induced apoptosis by c-Myc-dependent Bim activation. miR-34a overexpression resulted in sensitivity to cisplatin-induced apoptosis in U2OS cells by activation of c-Myc-dependent Bim pathway. Taken together, these results suggest miR-34a induction as an indicator of the therapeutic efficacy. They also provide an anticancer mechanism of miR-34a overexpression, and imply one of the potential strategies contributing to chemotherapeutic resistance in tumors.
Footnotes
Acknowledgments
Shandong Province College Science and Technology Project (J15LL10, ZR2015050013) and Shandong Natural Research Foundation (zr2016hm31) were received in support of this work.
Conflict of interest
The authors have no conflict of interest to report.