
Abstract
Keywords
Heavy menstrual bleeding (HMB), or menorrhagia, represents a frequent and clinically significant medical condition encountered by gynecologists, primary care physicians and nurse practitioners. Defined as regular, normal intervals of menstruation with volume exceeding 80 ml of blood loss per menstrual cycle [1], the impact of HMB on physical, social, emotional and/or material quality of life is the impetus for many women to seek treatment [2,3]. The use of therapies, such as hormonal medications, NSAIDs and surgical procedures, may reduce menstrual blood loss; however, concerns regarding suboptimal efficacy, side effects, and effects on fertility may limit their use [4].
For nearly four decades oral, immediate-release tranexamic acid, an antifibrinolytic, has been used in a myriad of applications, including the reduction of bleeding in surgical procedures and for treating women with HMB [5,6]. This formulation of tranexamic acid is used extensively worldwide and is generally well tolerated, with the majority of reported side effects being gastrointestinal (GI) in nature [5,6]. A novel, modified-release oral formulation of tranexamic acid (TA, Lysteda™, Ferring Pharmaceuticals Inc., Parsippany, NJ, USA) was recently approved by the US FDA for treating cyclic HMB. This modified-release formulation was designed to provide controlled dissolution of tranexamic acid into the stomach [7]. Phase III data for this novel formulation have demonstrated clinically significant improvements in menstrual blood loss and health-related quality of life aspects, with a low incidence of GI side effects across three [8], six [9] and 27 menstrual cycles [10]. In this article, we present an analysis of safety findings across nine menstrual cycles from an open-label extension study of the short-term (three- and six-cycle), Phase III studies of TA [8,9] in women with cyclic HMB.
Methods
Patients
Women, aged 18–49 years, with a history of cyclic HMB who completed a double-blind trial in one of the two pivotal efficacy studies of TA [8,9] and participated in all scheduled evaluations with no major protocol violations or study events were eligible for inclusion in this multicenter, open-label extension study at 76 clinical sites in the USA.
In the antecedent trials, patients met the inclusion criteria for HMB defined as an average menstrual blood loss of ≥80 ml/cycle during two pretreatment cycles, history of at least 6 months of regular menstrual cycles in normal range (21–35 days), menstrual periods lasting no more than 10 days, normal findings on pelvic examination, no clinically significant results on cervical cytology screens and no abnormal findings on transvaginal ultrasonograms.
Patients with a history or presence of clinically significant disease, anovulatory dysfunctional uterine bleeding, metrorrhagia, menometrorrhagia, polymenorrhea, endometrial polyps, endometrial hyperplasia, endometrial carcinoma, cervical carcinoma, myocardial infarction, ischemic disease, cerebrovascular accident, stroke, transient ischemic attack, thrombosis, thromboembolic disease or coagulopathy were excluded from the antecedent studies. Further exclusion criteria included abnormalities noted at physical examination; abnormalities on 12-lead ECGs; laboratory tests suggestive of a potential pituitary-prolactin stimulating tumor, uncontrolled hypothyroidism or severe anemia; and patients with a history of bilateral oophorectomy or who were pregnant, breastfeeding or planning to become pregnant.
Study design
Women who enrolled in this multicenter, open-label, extension study received 1.3 g TA orally three times daily for up to 5 days per menstrual cycle for a total of nine menstrual cycles. Participants were instructed to begin treatment at the onset of HMB and take the doses at least 6 h apart. Study visits were scheduled at 3-month intervals, immediately following treatment of the third, sixth, and ninth menstrual cycles. A follow-up phone call occurred 30 days (range: 25–35 days) after the last day the study drug was administered.
The use of anticoagulants, aminocaproic acid, hydroxychloroquine, hormonal contraceptives or herbal remedies was not allowed during the study. NSAIDs, acetaminophen, opioids, aspirin, cyclooxygenase-2 inhibitors, vitamins and iron therapy were permitted. At the discretion of the investigator, oral iron therapy could be initiated for patients exhibiting hemoglobin concentrations of 12 g/dl or less at enrollment, or patients who experienced a decline in hemoglobin to less than 11 g/dl while on study treatment. Patients with hemoglobin concentrations less than 8 g/dl or a relative reduction of hemoglobin greater than 3 g/dl while on therapy were withdrawn from the study.
The study was conducted from April 2007 to May 2009. The study received Institutional Review Board approval at each site and was conducted in accordance with the Declaration of Helsinki and is consistent with Good Clinical Practice and applicable regulatory requirements. All patients provided written informed consent.
Measures
Safety was the primary end point of this study. Safety was assessed by the incidence of treatment-emergent adverse events (AEs), findings on physical and gynecological examinations, vital sign measurements, laboratory test results (hematology, blood chemistry and urinalysis), ophthalmologic examinations (acuity, color blindness, intraocular pressure and dilated fundoscopic examination) and 12-lead ECGs. AEs were recorded throughout the study by spontaneous report by participants in patient diaries. These AEs were coded using Version 7.1 of the Medical Dictionary for Regulatory Activities (MedDRA®, Northrop Grumman Corporation, Los Angeles, CA, USA) and summarized using MedDRA-preferred terms within system organ class, severity, and relationship to treatment. The severity and causal relationship of AEs to study drug were assessed by the investigators based on clinical judgment. AEs were recorded from the time informed consent was signed until the participant completed the study. Vital sign and weight measurements were collected at all study visits and other safety observations, including ophthalmic, physical, gynecological examination findings, laboratory tests and ECGs were collected at the end point. Transvaginal ultrasonography and endometrial biopsy were performed if there was an abnormal finding on any gynecologic examination.
Statistical analysis
The sample size of the study was determined to ensure that at least 200 women would receive the study drug for 1 year or longer between the antecedent trials and current study. Safety analyses were conducted on the intent-to-treat (ITT) population (all study participants who received at least one dose of study drug). The summaries for quantitative variable included appropriate descriptive statistics (mean, standard deviation, median, minimum and maximum values). Safety data analyses were conducted using statistical software (SAS® Version 9.1.3, SAS Institute Inc., Cary, NC, USA).
Results
There were 311 women potentially eligible to enroll in the open-label extension study. Of these, 93% (288 out of 311) were enrolled. There were 28 women who failed to ingest at least one dose of study medication; therefore, 260 women comprised the ITT population
Patient disposition.

The number of patients enrolled was used as the denominator for calculated percentages.
The number of patients withdrawn was used as the denominator for calculated percentages.
Other events included irregular or discontinued menses, loss at follow-up, noncompliance with protocol, withdrawal of consent, elective surgery or intrauterine device placement and study site closure.
Protocol violations included taking a prohibited medication and not maintaining the study visit schedule.
ITT: Intent-to-treat.
Baseline and demographic characteristics were similar among participants. In the two pivotal efficacy study ITT populations, the median age of participants was 39.0 and 41.0 years, respectively, and the majority of women were white (64.2 and 73.5%, respectively). HMB in these populations was typically long-standing, with a median duration of 7.0 and 10.0 years, respectively.
Throughout this study, participants accumulated 6781 days of TA exposure, with a mean per patient exposure of 26.1 days in this nine-cycle study. The mean daily dose of TA was 3.8 g/day and the mean number of treatment days per cycle was 3.5 days. There were 1956 menstrual cycles in which at least one dose of study drug was taken. During the study, a large proportion of patients, 96% (249 out of 260), 81% (210 out of 260) and 65% (170 out of 260), received at least one dose of study drug for three, six and nine menstrual cycles, respectively. The most common concomitant medications taken during study medication dosing were anilides (i.e., acetaminophen) and propionic acid derivatives (i.e., ibuprofen, naproxen and ketoprofen).
Safety
During the 9 months of treatment with TA, 218 (83.9%) study participants experienced at least one treatment-emergent AE (i.e., an AE that occurred after receipt of at least one dose of study drug, regardless of causality). The majority of treatment-emergent AEs were of mild (175 out of 260, 67.3%) or moderate (154 out of 260, 59.2%) intensity
Treatment-emergent adverse events by severity occurring in ≥5% of patients.
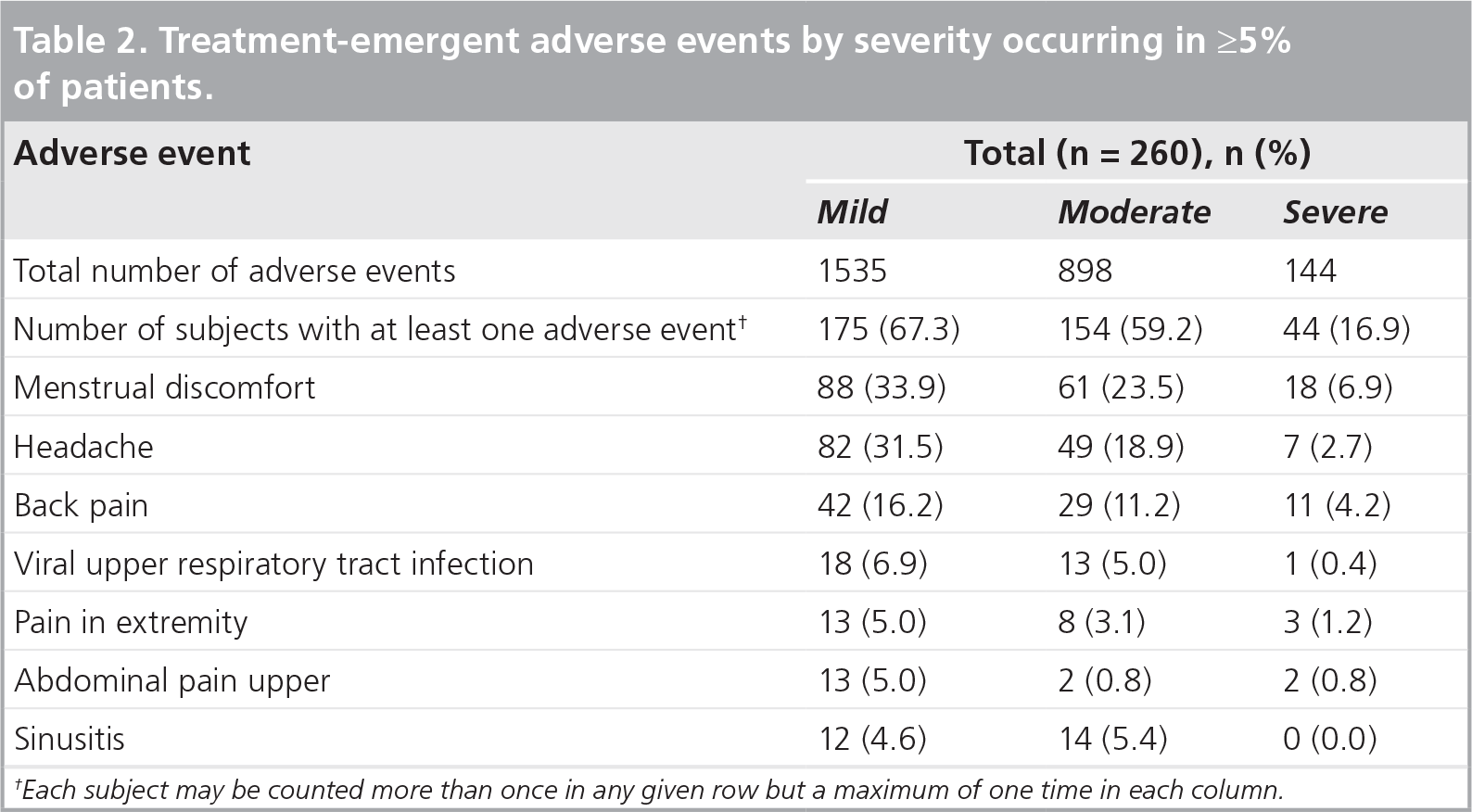
Each subject may be counted more than once in any given row but a maximum of one time in each column.
Treatment-emergent adverse events that occurred in ≥5% of patients.

Menstrual discomfort includes, but is not limited to, menstrual cramps, breakthrough bleeding, menstrual pain and spotting.
A total of five (1.9%) patients experienced serious AEs: a life-threatening decrease in blood glucose levels, a severe carcinoid tumor of the stomach, a life-threatening brainstem infarction, a life-threatening intracranial aneurysm, and a moderate trigeminal neuralgia. In addition, two patients reported serious AEs of menorrhagia (one of mild and one of moderate intensity). Each of these was considered to be not related to study drug by the investigator (either probably not [n = 5] or definitely not related [n = 2]). All but two patients (one patient who experienced a life-threatening decrease in blood glucose levels and one patient who experienced worsening of HMB) were withdrawn from the study. All serious AEs leading to study discontinuation resolved with the exception of the carcinoid tumor of the stomach, the intercranial aneurysm and the brain stem infarction, which were ongoing at the time of withdrawal.
In addition to the serious AEs that led to study discontinuation, three patients experienced AEs that resulted in withdrawal from the study. Of these, two patients withdrew due to three AEs considered to be definitely not or probably not related to study drug (one report each of decreased hemoglobin levels, menometorrhagia and menorrhagia) and one patient withdrew due to two AEs considered to be possibly related to study drug (one report each of dyspnea and throat tightness). All AEs leading to study discontinuation resolved with the exception of decreased hemoglobin levels, an issue that was ongoing at the time of withdrawal.
Gastrointestinal
Approximately 33% of participants (85 out of 260) reported GI treatment-emergent AEs during the study. Only 6.2% of patients (16 out of 260) reported GI treatment-emergent AEs that were considered by the investigator to be possibly related to the study drug, and there were no GI events probably or definitely related to study drug. The majority of GI events were of mild or moderate intensity; 3.5% of patients (9 out of 260) experienced GI-related events of severe intensity, including diarrhea, toothache, constipation, vomiting, nausea, abdominal discomfort and upper abdominal pain. The most frequently reported treatment-related GI events were upper abdominal pain (five women, 1.9%), diarrhea (five women, 1.9%), abdominal discomfort (four women, 1.5%), nausea (four women, 1.5%), vomiting (four women, 1.5%) and dyspepsia (two women, 0.8%).
Ophthalmic
Approximately 4% of patients (10 out of 260) reported ophthalmic AEs during the study. All eye disorder events were considered to be probably not or definitely not related to study drug with the exception of one report of blurred vision and one report of retinal artery stenosis. Most events occurred in one patient each and all were of mild (corneal pigmentation, dry eye, eye discharge, ocular hypertension, retinal artery stenosis, retinal pigmentation, eye pruritus and chalazion) or moderate (conjunctivitis and eye irritation) intensity. One event (blurred vision) occurred in two patients and both reports were of mild intensity. A patient experienced two separate episodes of blurred vision; the first event resolved without treatment intervention and the second event was ongoing at the time of study completion. There were no abnormalities noted in an ophthalmic examination performed approximately 1 month before the patient completed the study. In another patient, a retinal artery stenosis was noted on the ophthalmic examination during the final study visit. The patient did not receive any treatment for the event, which was considered stable and not clinically significant. No patient experienced eye disorder treatment-emergent AEs that were related to abnormal color vision.
There were no clinically significant results from the ophthalmic examinations. Four patients were noted to have high intraocular pressures. One of these patients experienced ocular hypertension that was recorded as an AE considered by the investigator to be probably not related to the study drug and was ongoing after completion of the study. No patient withdrew from the study as a result of high intraocular pressure.
Cardiovascular
No thrombotic or thromboembolic treatment-emergent AEs occurred during the study. Overall, there was no evidence of prolongation of QTc intervals because of treatment with TA. Three patients had elevated QTcF values during the course of the study. No cardiovascular-related AEs were reported in these subjects leading to study discontinuation; however, one patient withdrew early from the study due to noncompliance with the protocol. A treatment-emergent AE of nonspecific ST–T wave changes was reported in one patient; this event was of mild intensity, considered probably not related to study drug and was resolved without intervention.
Physical/gynecological
Patients with abnormal results considered to be clinically important at physical or gynecologic examinations had AEs to reflect the finding. Three patients had AEs (one report each of salpingitis, uterine leiomyoma, endometrial thickness and ovarian cyst) that corresponded with abnormalities noted on transvaginal ultrasonograms. All of these events were of mild or moderate intensity and were considered by the investigator to be probably not or definitely not related to the study drug. No patient discontinued the study as a result of these AEs. No pregnancies were reported during the study.
Vital signs
After nine cycles of treatment, systolic and diastolic blood pressure, pulse rate and weight measurements in patients were similar to those reported in patients after three cycles. Five patients experienced AEs related to vital sign measurements: one report each of weight increase and hypotension and three reports of hypertension. All these events were of mild-to-moderate intensity and, with the exception of weight increase, all other vital sign-related events were considered probably not related to study drug. No patient withdrew from the study as a result of these AEs.
Laboratory parameters
An AE associated with an abnormal finding on laboratory tests (hematological, clinical chemistry or urinalysis) was experienced by 44 (16.9%) patients. In these abnormal test results in which AEs were corroborated, the AEs were considered to be definitely not or probably not related to study drug, with the exception of five events occurring in four patients that were considered to be possibly related to study drug (two reports of elevated liver enzymes in one patient and one report each of anemia, trace occult blood in urine and positive occult blood in urine). All related treatment-emergent AEs resolved without intervention and no patient discontinued as a result of these events. The patient who experienced positive occult blood in urine did not have any treatment recorded for the event and withdrew from the study because she moved from the area.
Discussion
A novel, oral formulation of TA was observed to be safe and well-tolerated in women with cyclic HMB in this open-label, nine-cycle extension study. The results are consistent with the established safety profile of tranexamic acid [5,6]. During the study, nearly two-thirds of women received at least one dose of TA during nine cycles of extended treatment. Although patients were allowed 5 consecutive days of treatment per cycle, mean TA exposure was less than 4 days per cycle. These findings are consistent with the observation that the majority of menstrual blood loss occurs during the first 3 days of menstruation [11]. Less than 10% of patients withdrew from this 9 month study because of an AE, similar to patient discontinuation rates noted in other studies of tranexamic acid [6,8,9]. Less than 2% of women reported serious AEs, and nearly all of these events were considered unrelated to the study drug. The incidence of GI-related AEs reported during the study was low and similar to rates reported in Phase I and Phase III studies of modified-release tranexamic acid [12].
Ophthalmic disorders were closely monitored in this study due to preclinical findings of retinal degeneration with higher doses of tranexamic acid than used within the current study and other studies using TA, as well as previous reports of color vision or visual acuity disturbances in patients treated with tranexamic acid [5,13]. Although the etiology has yet to be determined, visual changes reported in the literature have typically been transient and resolved after drug cessation [13]. In this study, the only ocular-related finding was high intraocular pressure, which did not result in significant ophthalmic impairment. Although there were no ophthalmic AEs related to abnormal color vision, there were two AEs of blurred vision and one AE of retinal artery stenosis.
During more than four decades of use worldwide, there have been periodic assertions of increased thrombotic risk with tranexamic acid [14]. However, the lack of thromboembolic events in this study is consistent with larger-scale population risk estimates that demonstrate no increased risk with TA treatment [4,15]. Although tranexamic acid is known to decrease the fibrinolytic activity in the peripheral circulation, it has been postulated that the preservation of fibrinolytic activity in the vascular wall may explain the nonthrombogenic effect of tranexamic acid [4]. Moreover, reports have suggested that the rate of incidence for developing a thromboembolic event in patients treated with tranexamic acid is similar to that reported spontaneously within the general population [15,16]. Furthermore, an increased risk of thromboembolism has been associated in women with HMB who are diagnosed with anemia, which may lend to speculation that HMB is a prothrombotic condition [14].
In comparison to the three- and six-cycle double-blinded clinical trials of TA, the use of NSAIDs was permitted during dosing in this study. As NSAIDs are among the most common medications in the USA, and for women, they are often used to relieve menstrual discomfort [17], there is a likelihood for the concomitant use of NSAIDs and TA in the general population. Additionally, certain NSAIDs such as mefanamic acid, naproxen, ibuprofen and flurbiprofen have been investigated as treatment options for HMB, although their efficacy in reducing menstrual blood loss is generally low [18]. In this study, the use of NSAIDs during TA dosing did not adversely affect the safety or tolerability of TA.
In the present study, a large proportion of the ITT population, almost 75%, completed the nine treatment cycles with TA. Failure to return was the predominant reason for study withdrawal. The authors note that the following could be potential reasons for not returning to study visits: relocation to a different city, scheduling conflicts, or an unwillingness to participate in the number of study visits and the number of study-related procedures, when compared with routine practice, for a long-term study.
Limitations
This study had several limitations. Due to the spontaneous nature of reporting AEs and the lack of a control, the true frequency of AEs associated with tranexamic acid compared with treatment-independent events is difficult to determine without a placebo comparator. Also, as the primary goal of the study was to assess safety, the study was not designed to evaluate the efficacy of TA. All patients who were eligible to continue into the open-label extension study completed one of two double-blind treatments; therefore, patients who did not tolerate treatment or were dissatisfied with efficacy during double-blind treatment would not be expected to have opted into the present study.
Although the use of NSAIDs was allowed, the prohibition of medications such as hormonal contraceptives may not present a complete picture of a real-world treatment scenario. Additional data are needed to determine if concomitant use of these medications adversely affect the safety or tolerability of tranexamic acid. However, as TA is usually effective in treating cyclic HMB and combination oral contraceptives are known to be associated with increased thrombotic risks, many clinicians do not commonly prescribe these therapies together.
Conclusion
In this extension study of nine menstrual cycles, TA at 3.9 g/day was generally well tolerated when used for up to 5 days and exhibited a favorable safety profile. TA is an oral, nonhormonal, non-surgical alternative for intermittent use in women with cyclic HMB.
Future perspective
Given the applicability of immediate-release tranexamic acid in other indications, we welcome studies that include modified-release tranexamic acid for surgical procedures, hemorrhages, dentistry and hemophilia. We also look forward to the impending results of the pharmacokinetic assessment of modified-release tranexamic acid in an adolescent population with evidence of HMB.
Executive summary
The goal of this multicenter, open-label, extension clinical study was to assess the safety of a novel, oral formulation of tranexamic acid (TA, Lysteda™) in women with cyclic heavy menstrual bleeding across nine menstrual cycles.
This novel, modified-release oral formulation of TA was designed to provide controlled dissolution of tranexamic acid into the stomach.
Safety was assessed by the incidence of spontaneously reported adverse events (AEs), findings on physical and gynecological examinations, vital sign measurements, laboratory test results, findings on ophthalmologic examinations and findings on ECGs.
Frequently reported AEs were consistent with those observed with the use of an immediate-release formulation of tranexamic acid and included menstrual discomfort, headache and back pain.
The majority of treatment-emergent AEs were of mild or moderate intensity.
The incidence of treatment-related gastrointestinal treatment-emergent AEs was low across nine cycles of treatment.
There was no evidence of ocular toxicity or prothrombotic effects during treatment with TA.
No patients discontinued the study as a result of abnormal findings on physical and gynecological examinations, vital sign measurements, laboratory tests or findings on ECGs.
During a long-term treatment study, treatment with TA was well tolerated when used for up to 5 days during menstrual cycles and was consistent with the established safety profile.
This formulation of TA adds an oral, nonhormonal therapeutic option for use to the armamentarium available to treat cyclic heavy menstrual bleeding.
Footnotes
Acknowledgements