
Abstract
The aim of this research is to investigate sodium perborate tetrahydrate (SP) modification of cellulosic fibers. Flax, jute, and sisal fibers were treated with aqueous solutions of SP at three different concentrations. The changes in surface characteristics were analyzed using Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), X-ray difraction (XRD), thermogravimetric analysis (TGA), and scanning electron microscopy (SEM). No significant changes in the crystallinity index for all fibers tested were observed as SP treatments only produced surface modification. SEM images showed partial removal of non-cellulosic components occurred. Increased SP concentrations led to greater surface cleaning and fiber separation, imparting a more hydrophobic surface character.
Introduction
Lignocellulosic and/or pectocellulosic fibers are naturally-occurring resources available all over the earth's tropical and temperate regions. 1 The past 20 years have witnessed a high demand for plant fibers as reinforcements for polymeric composites. 2 With their low cost, sustainability, recyclability, and reduced tool wear during manufacturing processes, these fibers are increasingly gaining attention as reinforcements in composite materials for various end uses, including civil engineering and transportation related applications.
Cellulosic fibers are generally classified into four groups— bast, leaf, seed, and fruit fibers—that can be used for reinforcement and/or as additives for polymer-based materials. The principal chemical constituents of plant fibers are cellulose, hemicelluloses, and lignin. Plant fibers also contain pectin, waxy substances, fats, inorganic matter, nitrogenous matter, and traces of pigments. 3 These contents can vary with the type of plant—as well as the part of the plant where the fibers are taken from—the growing region, and the growing season.
The presence of impurities and non-cellulosic components on the surface of cellulosic fibers, as well as their highly polar nature imparted by a large number of hydroxyl groups, hinder mechanical interlocking and cause incompatibility with non-polar matrices. Many studies have been reported on the modification of cellulosic fiber surface characteristics for use as reinforcement materials in composites.4-7 To obtain polymeric composite materials reinforced with cellulosic fibers, interfacial bonding between the fiber and the polymer must occur. Development of a good interface depends on the fiber and the polymeric matrix being used. 1 Both components of the composite material are usually modified chemically or physically.8-13 Physical modifications include corona or plasma treatment and steam explosion 14 whereas cellulosic fibers are chemically modified by alkali, silane coupling agents, graft copolymerization, dilute polymer solutions, and oxidizing agents.15-19 The use of oxidizing agents for modifying the surface characteristics of cellulosic fibers by increasing surface hydrophobicity and surface roughness was previously reported.20,21
Sodium perborate (SP), a source of active oxygen, can produce hydrogen peroxide oxidizing agent when dissolved in water. SP can produce perhydroxide ions that can hydrolyze derivatives of carboxylic acids to form active peroxylate moieties under basic conditions. This reaction occurs at a relatively lower temperature than that for the formation of hydrogen peroxide. 22 Oxidation reactions of hydrogen peroxide in alkaline medium are given in Eqs. 1–7. 23

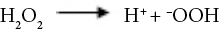

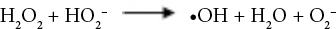

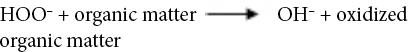
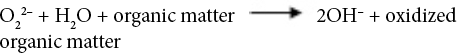
SP can be a good alternative agent for surface treatment of cel-lulosic fibers. However, a limited number of studies using SP to modify cellulosic fibers were performed.24,25 Bulut and Aksit stated that SP treatment provided the greatest interfacial adhesion with polypropylene by increasing the surface roughness and surface carbon to oxygen ratio (C/O) of the jute fibers when compared to other commonly-used oxidative agents. 24 Seki et al. noted that an optimal concentration of SP was used to enhance the surface hydrophobicity of jute fibers. 25
The aim of this current research is to assess SP treatment on the surface characteristics of bast fibers such as sisal (leaf fiber), flax, and jute. To properly evaluate changes in the fiber surface and structure after treatment, Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectros-copy (XPS), X-ray diffraction (XRD), and thermogravimetric analysis (TGA) were performed. The fibers were also studied by scanning electron microscopy (SEM).
Experimental
Materials and Fiber Treatment
The flax, sisal, and jute fibers used in this study contained 70, 76.0, and 63.5% cellulose, respectively, and were provided by local suppliers. The cellulosic contents of the fibers were determined in accordance with Erdogan et al. 26 For fiber treatment, SP tetrahydrate (NaBO3·4H2O) was provided by Merck Corp. Prior to treatment of the fibers, jute (J), sisal (S), and fax (F) fibers were soaked in beakers containing distilled water for 1.5 h to remove surface impurities and then oven-dried until totally dry to remove free water from the fibers. The fibers were then immersed in 2% (J2, S2, F2), 5% (J5, S5, F5), and 10% (J10, S10, F10) w/v SP aqueous solutions at 75 °C for 30 min. 25 After chemical processing, the fibers were removed and rinsed with distilled water several times to remove the chemical residues. The fibers were oven-dried until totally dry at 105 °C.
FTIR Analysis
FTIR spectra of the untreated and treated fibers were obtained by FTIR spectroscopy (Perkin Elmer Spectrum BX). Spectra were recorded in the range of 600-4000 cm−1 with a resolution of 2 cm−1.
XPS Analysis
Surface chemistry of the fibers was analyzed by XPS (Ther-moScientific) with a monochromatic Al-Kα (1486.7eV) X-ray source and a 400-nm diameter beam size. The vacuum pressure was below 10−9 Torr during spectral data acquisition. XPS data were acquired between 1350 and 10 eV applying a pass energy of 150 eV and a resolution of 1 eV. A total of 15 scans from a random single point were recorded. The sample surface for each of the fibers tested were sputtered with ionic Ar gas before operation.
XRD Analysis
The X-ray diffraction pattern for the samples was obtained by a Rigaku ULTIMA 3-Rint 2200/PC system with Cu-Kα radiation. The diffraction intensities of the fibers were recorded in the 3 to 60° range at 40 kV and 36 mA with a scan speed of 2°/min. To determine the effect of modification on the fibers’ crystallinity index (CI), the empirical formula proposed by Segal et al. was used (Eq. 8).27,28
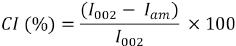
I002 is the maximum intensity of the diffraction peak the (002) lattice peak at a 2θ angle between 22° and 23°, which represents both crystalline and amorphous materials. Iam is the minimum intensity of the diffraction representing the amorphous materials, which is taken at a 2θ angle between 18° and 19°.28,29
TGA
Thermal decomposition behavior of the fibers were studied by TGA using a Shimadzu DTG-60H instrument by heating from room temperature (RT) to 700 °C with a heating rate of 10 °C/min under a dry nitrogen atmosphere. An alumina (Al2O3) pan was used as a container and reference material.
Surface Morphology
Surface morphology of the untreated and treated fax, jute, and sisal fibers were analyzed using a JEOL-JJM 6060 SEM. SEM micrographs were obtained with accelerating magnifications of 500× to 3000×.
Results and Discussion
FTIR Analysis
The functional groups of the untreated and treated fibers were analyzed through FTIR-attenuated total reflectance (ATR) analysis. The absorption bands corresponding to the functional groups of the fibers are tabulated in Table I. FTIR spectra of the untreated and treated fibers were recorded from 4000 to 600 cm−1. The characteristic features of the spectra were due to the cellulose, hemicellulose, lignin, and pectin constituents of the cellulosic fibers. 30 A strong absorption band was observed at 3339 cm−1 for all untreated test fibers due to O-H stretching and hydrogen bonds. 31 The peak at 2917 cm−1 was due to the C-H stretching vibration from CH and CH2 groups in cellulose and hemicellulose. 32 The peaks at 1734 and 1249 cm−1 can be attributed to C=O and C-O stretching vibrations of hemicellulose, respectively. 33 The band at 1617 cm−1 was assigned to the antisymmetric COO− stretching or to the presence of water. 33 The absorptions at 1500 to 1600 cm−1 corresponded to the C=C of aromatic skeletal vibrations in lignin, which decreased after modification. 34 The C-O-C stretching of side substituents of xylan and lignin aromatic C-O stretching were located at around 1030 cm−1. 30 The small peaks from 1155 to 1165 cm−1 corresponded to C-O-C glycosidic bonds from the polysac-charide component, which was largely cellulose. 30
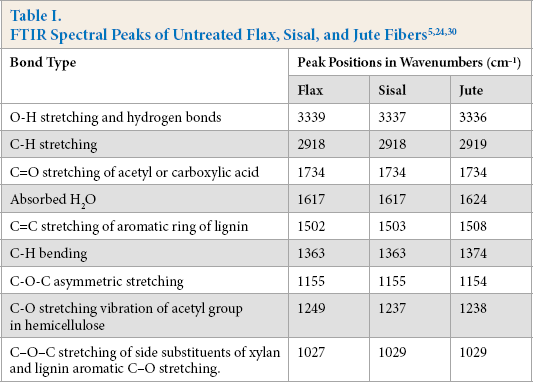
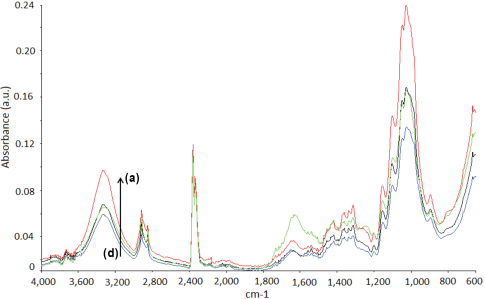
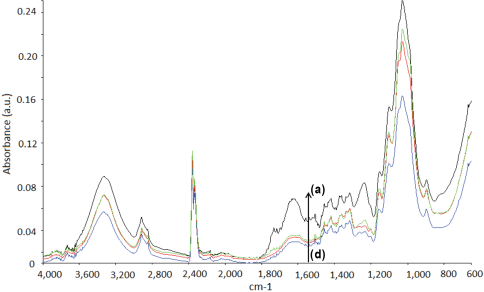
FTIR spectra of the untreated and treated fax, jute, and sisal fibers are given in Figs. 1–3, respectively. The intensity of the peaks centered in the 3340 and 2919 cm−1 regions indicated O-H stretching and C-H stretching changes after treatment, respectively. The peaks at 1734 and 1245 cm−1, assigned to the C=O stretching of acetyl or carboxylic acid and C-O stretching vibration of the acetyl group in hemicellulose, showed diminishing intensities after fiber treatment. 35 This may be due to partial removal of surface impurities. Additionally, the absorptions in the 1500 to 1600 cm−1 region, corresponding to the aromatic C=C skeletal vibrations of lignin, decreased after treatment for nearly all cellu-losic fibers. 34 Treatment resulted in notable changes in the intensity of the peak located at ∼1030 cm−1. This may be due to the changes in the amount of xylan with aldehyde/acid/carboxylate substituents. 30
FTIR spectra of flax fibers. (a) F2, (b), untreated fax, (c) F5, and (d) F10.
FTIR spectra of jute fibers. (a) Untreated jute, (b) J2, (c) J10, and (d) J5.
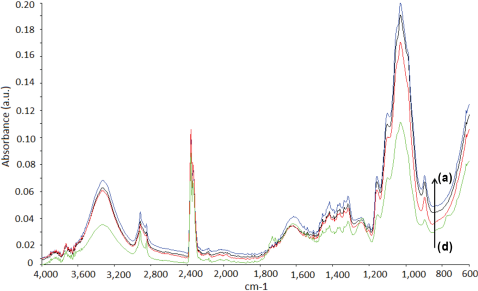
FTIR spectra of sisal fibers. (a) S10, (b) S5, (c) S2, and (d) untreated sisal.
Fiber Surface Chemistry
The surface chemistry of the untreated fibers and the fibers treated with 10% SP solution was analyzed using XPS. Table II gives the percent carbon and oxygen contents and the C/O ratio of the fiber surfaces. The oxygen and carbon contents of the untreated fibers changed after SP modification (Table II). Carbon contents increased, while oxygen contents decreased after treatment. The C/O ratios for the untreated fax, jute, and sisal fibers were 3.59, 3.80, and 3.43, respectively. For the 10% SP treated fibers, the C/O ratios of F10, J10, and S10 fibers were 6.14, 5.60, and 5.48, respectively. The C/O ratio can be used to characterize the surface polarity of materials.24,36 These results showed that the 10% SP treatment improved the surface hydrophobicity of the cellulosic fibers studied.
XPS Fiber Analysis a
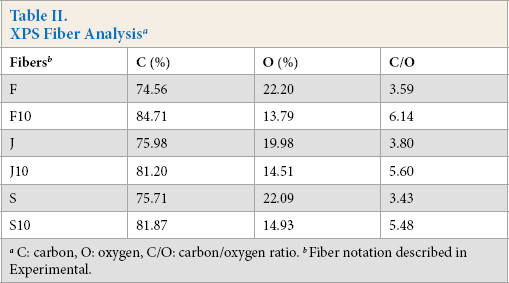
C: carbon, O: oxygen, C/O: carbon/oxygen ratio.
Fiber notation described in Experimental.
Fiber Crystallinity
Fig. 4 shows the XRD patterns of the untreated and treated fibers. The untreated fibers gave main diffraction peaks at 2θ = 22-23° (002) and 2θ = 16-17° (110), which are typical diffractions of cellulose I. 33 Flax fibers had the highest CI values. Jute and sisal fibers had similar CI values (Table III). This is due to the amounts of cellulosic and non-cellulosic components of these fibers. The cellulosic contents of fax, jute, and sisal fibers used in this research were 70, 63.5, and 76%, respectively. 37 Accordingly, the fiber with the greater cellulose content gave greater CI values. It is notable that the XRD patterns of the untreated and treated fibers resembled each other, with only slight changes in the intensities of the diffraction peaks. The untreated and treated fibers presented similar CI results. This may indicate the surface modification effect of the SP treatment.
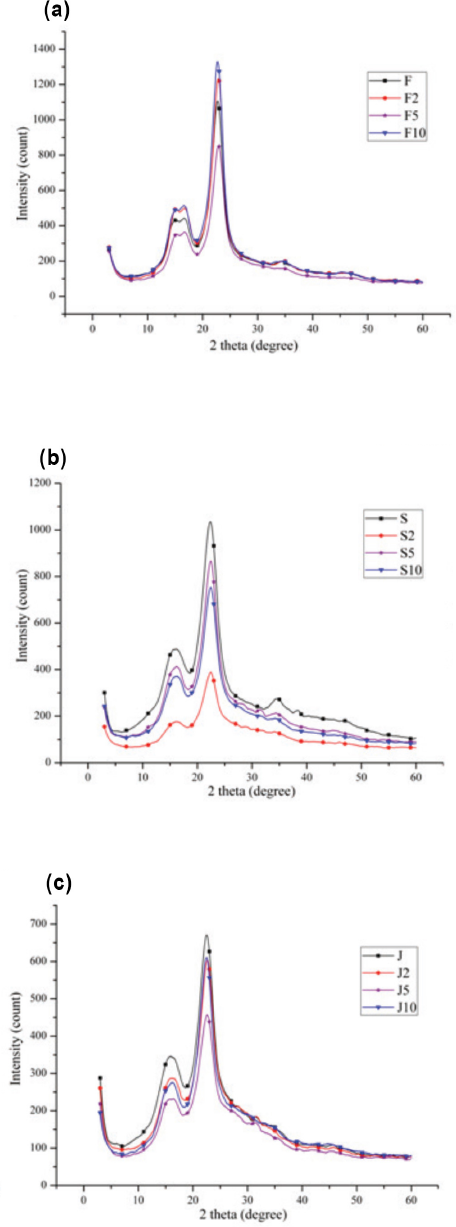
XRD patterns of untreated and treated fibers. (a) Flax, (b) sisal, and (c) jute.
Crystallinity Indexes (CI) of the Fibers
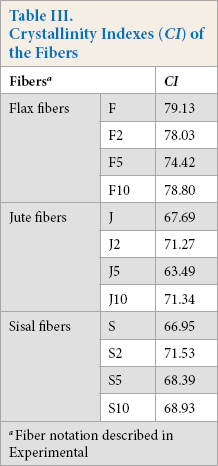
Fiber notation described in Experimental
Fiber Thermal Stability
Table IV lists the thermal decomposition data of the untreated and 10% SP treated fibers. Fig. 5 presents the TGA and derivative TGA curves for the fibers. The mass losses at ∼120 °C indicated that the moisture content of the fibers were similar and in the range of 4.71 to 7.52%. The untreated fibers had two distinct degradation slopes, whereas the treated fibers showed only one (Fig. 5). The first slope in the temperature range of 250 to 320 °C was due to hemicellulose degradation. 38 This may explain why the treated fibers showed one slope. Hemicellulose molecules are soluble polysaccharides in water and alkali solutions, 39 such as SP solutions.
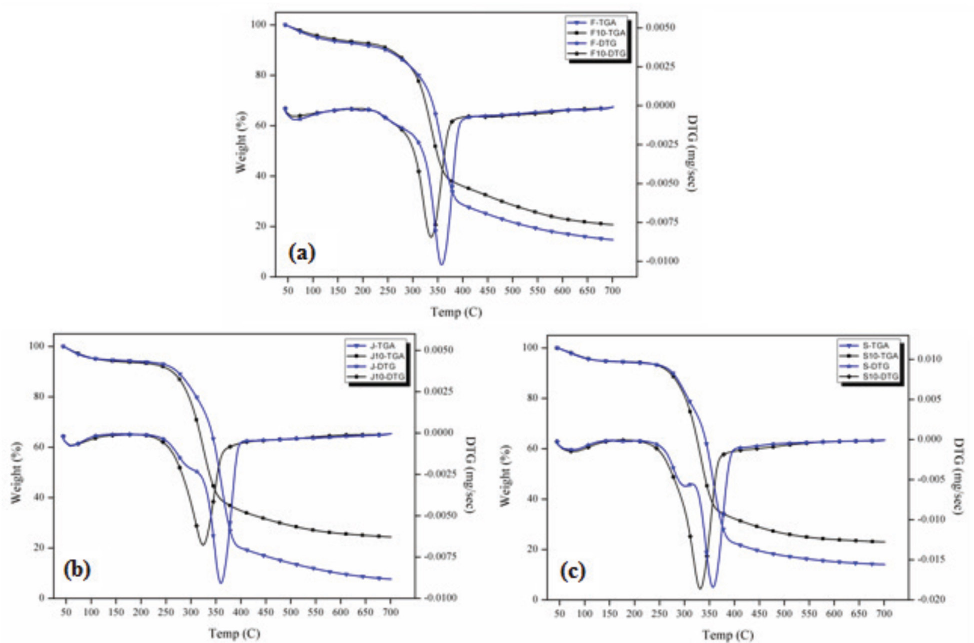
TGA curves of untreated and treated cellulosic fibers. (a) Flax, (b) jute, and (c) sisal.
TGA Data for the Fibers
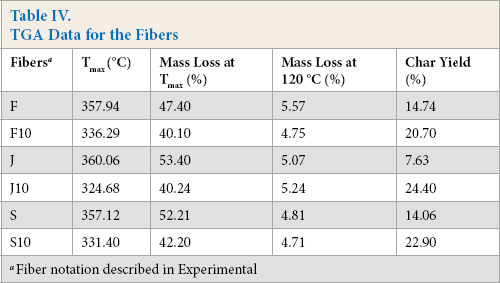
Fiber notation described in Experimental
Cellulose degradation, which is the second step of untreated fiber degradation and the only step of treated fiber degradation, occurred between 260 and 400 °C. In this degradation step, the maximum degradation temperature (Tmax) of the treated fibers decreased significantly as a result of the treatment. This may be due to the partial removal of lignin after treatment, which is in agreement with the FTIR results (Figs. 2–5). The intensity of the peaks detected in the range of 1502 to 1508 cm−1 (aromatic C=C skeletal vibrations in lignin) decreased after treatment. Lignin carbons are more stable than those in cellulose due to lignin's higher aromatic con-tent. 40 Table IV shows that the char yield values of the fibers increased remarkably after treatment. This may indicate more favorable thermal stability.
SEM Analysis
SEM images of the untreated and treated sisal, fax, and jute fibers are presented in Figs. 6–8, respectively. As confirmed from SEM images of the untreated fibers, these fibers exist as bundles of unit cells aligned longitudinally and bound firmly together with lignin rich, weak intermolecular bonds. 2 The surfaces of the untreated fibers are covered with impurities and non-cellulosic components.41,42
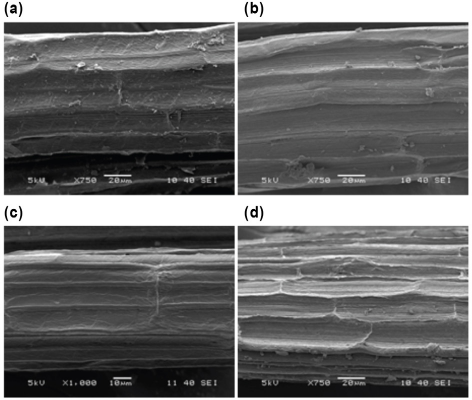
SEM images of (a) S (×750), (b) S2 (×750), (c) S5 (×1000), and (d) S10 (×750).

SEM images of (a) F (×2000), (b) F2 (×2000), (c) F5 (×3000), and (d) F10 (×3000).
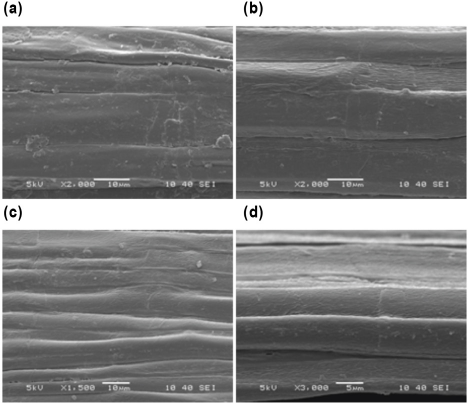
SEM images of (a) J (×2000), (b) J2 (×2000), (c) J5 (×1500), and (d) J10 (×3000).
Sisal fibers are formed as discontinuous, short individual cells joined together end-to-end apart from the other fibers.2,43 The treated fibers showed more opened cells of the fibers and bundles of microfibrils with increased SP concentration. The surface topography of the treated sisal fibers became rougher after treatment, which is desirable for fiber reinforced composite materials. For fax and jute fibers, partial removal of the non-cellulosic components and separation of the elementary fibers occurred as a result of the treatment.
Conclusion
This work was a comparative study on SP modified jute, fax, and sisal fibers at various SP concentrations. Chemical characterization showed dramatic increases in the C/O ratio on fiber surfaces after 10% SP treatment. This can provide greater hydrophobic surface characteristics beneficial to composite manufacturing. SP treatment did not obviously alter the CI value of the fibers. The surface of the fibers after treatment were cleaner and rougher due to the partial removal of impurities and bonding materials between the fiber elements. The maximum thermal degradation temperatures of the fibers decreased, owing to the partial removal of lignin after SP treatments, but char yield values of the fibers showed apparent increases that could benefit composite material manufacturing.
Footnotes
Acknowledgements
The authors would like to thank to the Center for Fabrication and Application of Electronic Materials at Dokuz Eylul University for their valuable support.