
Abstract
In vitro and in vivo experimental studies suggest that the transcription factor NF-κB plays a role in tubulointerstitial injury. We investigated possible cellular and molecular mechanisms involving NF-κB activation in the progression of tubulointerstitial lesions in human lupus nephritis (LN). Paraffin-embedded renal biopsies from 50 patients with LN and six control patients with minimal change disease (MCD) were examined by Southwestern histochemistry for in situ detection of active NF-κB and AP-1. Immunohistochemistry was performed to examine the expression of NF-κB, AP-1, and NF-κB regulatory proteins (IκB-α, p-IκB-α, and IKK-α proteins), as well as NF-κB and AP-1 downstream target proinflammatory molecules (ICAM-1, TNF-α, IL-1β, IL-6, and GM-CSF) and NF-κB upstream signaling molecules (CD40 and CD40L). We observed extensive upregulation of activated NF-κB in renal tubular cells and interstitial cells, in parallel with overactivation of transcription factor AP-1 in LN, as compared with normal controls and MCD. Tubular expression of activated NF-κB correlated well with the degree of tubulointerstitial histopathological indices and/or renal function. Tubulointerstitial IKK-α expression was specifically upregulated in LN. IκB-α and p-IκB-α were detected only in interstitial cells in LN. Tubulointerstitial expression levels of NF-κB and AP-1 downstream inflammatory molecules and NF-κB upstream signaling molecules CD40 and CD40L were markedly enhanced in LN as compared with MCD or normal controls and were associated with tubulointerstitial histopathological indices and/or renal function. The results suggest that altered IKK-α expression and NF-κB activation along with AP-1 overexpression may play a pathogenic role in tubulointerstitial injury in human LN mediated through a network of downstream proinflammatory molecules.
Keywords
T
The transcription factor nuclear factor-κB (NF-κB) is a key regulator of the expression of numerous proteins involved in the inflammatory response (Li and Verma 2002). Many inflammatory molecules are involved in the progression of tubulointerstitial injury (Daha and van Kooten 2000) such as the adhesion molecules (e.g., ICAM-1/VCAM-1) and proinflammatory cytokines (e.g., TNF-α, IL-1, IL-6, and GM-CSF), which are known to be regulated independently and/or coregulated by transcription factor NF-κB and other transcription factors such as activated protein (AP-1) (Schreck and Baeuerle 1990; Sung et al. 1991; Newell et al. 1994; Thomas et al. 1997; Roebuck et al. 1999; Berendji-Grun et al. 2001; Guijarro and Egido 2001; Udalova and Kwiatkowski 2001). The classic form of NF-κB is a heterodimer of a p65 and p50 subunit in the kidney (Guijarro and Egido 2001). In the cytoplasm, NF-κB remains inactive through binding to its endogenous inhibitor known as inhibitor κB-α (IκB-α).A variety of stimuli can lead to the dissociation of this complex resulting in the release of active NF-κB. Degradation of IκB-α occurs through a mechanism by which it is phosphorylated by IκB kinase (IKK). The released NF-κB translocates to the nucleus where it may activate the transcription of downstream target genes (e.g., TNF-α and IL-1β), usually in collaboration with other transcription factors such as AP-1 (Ghosh et al. 1998).
NF-κB activation in renal tubular cells has been implicated in tubulointerstitial injury in proteinuria-induced rat models (Rangan et al. 1999; Gomez-Garre et al. 2001; Takase et al. 2003). In experimental proteinuric nephropathy characterized by tubulointerstitial injury, blocking NF-κB and AP-1 activation attenuated tubulointerstitial injury (Rangan et al. 1999; Takase et al. 2003). Gene transfer of truncated IκB-aαinto the tubulointerstitium markedly attenuated proteinuria-induced tubulointerstitial injury by specifically inhibiting tubular NF-κB activation along with tubular expression of NF-κB-dependent inflammatory mediators (Takase et al. 2003). Upregulation of activated tubular NF-κB has been suggested to play a role in tubulointerstitial injury, in parallel with upregulation of tubular expression of adhesion molecules, chemokines, or cytokines in human IgA nephropathy, membranous nephropathy (MN), minimal change disease (MCD), and diabetic nephropathy (Mezzano et al. 2001, 2004; Ashizawa et al. 2003). However, scarce data are available on the role of NF-κB activation in tubulointerstitial inflammation of LN.
Clinical and histopathological data of patients participating in the study
Biopsy samples were processed for routine analysis by light microscopy, immunofluorescence analysis, and/or electron microscopy for the diagnosis of LN as described previously (Khan and Sinniah 1995). Biopsies diagnosed as LN were classified according to the criteria defined by the International Society of Nephrology/Renal Pathology Society in 2003 (Weening et al. 2004). LN disease groups consisted of two subjects with focal LN (class III), 35 subjects with diffuse LN (class IV), and nine subjects with membranous LN (class V). The class IV group was comprised of five subjects with diffuse segmental LN (class IV-S) and 30 subjects with diffuse global LN (class IV-G).
Immunohistochemistry
The monoclonal mouse anti-RelA/p65 antibody (Chemicon International; Temecula, CA), which is directed against an epitope of p65 containing a nuclear localization signal, is specific for the detection of activated NF-κB (Kaltschmidt et al. 1995; Mezzano et al. 2001). Polyclonal goat anti-p50 and rabbit anti-c-jun and anti-c-fos antibodies (Santa Cruz Biotechnology; Santa Cruz, CA) were also applied on paraffin renal tissues previously by other groups (Mezzano et al. 2001). Polyclonal rabbit antibodies against human TNF-α, IL-1β (Rockland Immunochemicals; Gilbertsville, PA), and IL-6 (Pierce Biotechnology; Rockford, IL) and monoclonal mouse antibody against ICAM-1 (Santa Cruz Biotechnology) were used in our previous studies (Qiu et al. 2004; Zheng et al. 2006a). Other primary antibodies used were polyclonal rabbit anti-IKK-α, monoclonal mouse anti-IκB-α and -phosphorylated IκB-α (serine 32), and polyclonal goat anti-CD40L antibodies (Santa Cruz Biotechnology); polyclonal rabbit anti-GM-CSF antibody (Rockland Immunochemicals); monoclonal mouse anti-CD40 antibody (Serotect; Oxford, UK); and monoclonal mouse anti-CD68KP1 antibody (Dako; Carpinteria, CA). Specificity of other antibodies was confirmed by their reactivity with specific human targets by immunoprecipitation and/or Western blot analysis according to the manufacturer's description.
IHC was conducted on 4-μm-thick paraformaldehyde-fixed paraffin sections. Except for CD68, slides were subjected to microwave antigen retrieval with wet heat at 98C for 10 min at 150 W (Milestone Microwave Systems; Bergamo, Italy). Citrate buffer (100 mM, pH 6.0) was used with anti-p65, anti-p50, and anti-CD40L; 0.01 mol/liter, pH 6.0, citrate buffer was used with antibodies against c-jun, c-fos, IκB-α, p-IκB-α, IKK-α, CD40, TNF-α, IL-1β, IL-6, GM-CSF, and ICAM-1 before peroxidase blocking. Slides were predigested with protease for CD68 immunostaining. A two-step EnVision+ System peroxidase kit (Dako) as described previously (Zheng et al. 2006a) was used for the detection of p65, IKK-α, IκB-α, p-IκB-α, CD40, TNF-α, IL-1β, IL-6, GM-CSF, and ICAM-1. For anti-p50 and -CD40L, a three-step Dako LSAB+ Peroxidase Kit was used (Zheng et al. 2006a). DAB was used for colorimetric detection followed by mild counterstaining with PAS reagents and Gill's hematoxylin. Specificity of primary or secondary antibodies was verified by the replacement of primary antibodies with equal concentrations of non-immune sera or by omitting primary antibodies. Positive control was tonsillitis tissue for detection of these molecules.
SWH
SWH was performed as described previously (Hernandez-Presa et al. 1999). In brief, digoxigenin (DIG)-labeled double-stranded synthetic DNA with the consensus sequence of NF-κB (sense: 5′-AGTTGAGGGGACTTTCCCAGGC-3′) or with the consensus sequence of AP-1 (sense: 5′-CGCTTGATGAGTCAGCCGGAA-3′) (Gibco-BRL, Life Technology; Gaithersburg, MD) was used as a probe (DIG oligonucleotide 3′-end labeling kit; Roche Diagnostics, Indianapolis, IN).
Four-μM-thick paraffin-embedded kidney sections were dewaxed and rehydrated. Preparations were incubated with levamisole (Sigma; St Louis, MO) and postfixed with 0.2% paraformaldehyde for 30 min at 28C. Following pretreatment with pepsin A (433 U/mg; Sigma) in 1 N HCl for 30 min, sections were incubated with 0.1 mg/ml DNase I in HEPES–BSA for 30 min at 30C. Labeled probes diluted to 100 pmol/liter in HEPES–BSA containing 0.5 μg/ml poly(dI-dC) (Roche Diagnostics) were applied to each slide overnight at 37C. After incubation in blocking solution (0.01X SSC, 0.01% sodium dodecyl sulfate, 0.03% Tween 20, 0.1 mol/liter maleic acid, 0.15 mol/liter NaCl, pH 7.5), anti-DIG antibody conjugated with alkaline phosphatase (1:250 in blocking solution; Roche) was added overnight at 4C. Color reaction was developed using nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate (NBT/BCIP; Roche) according to the manufacturer's instructions. Slides were counterstained with PAS reagent minus hematoxylin.
The following conditions were carried out as negative controls: (1) absence of probe; (2) DIG-labeled mutant NF-κB (sense: 5′-AGTTGAGG
Semi-quantitative Histological Assessment
Histological evaluation was performed using light microscopy by two independent pathologists (LZ and RS) who were blinded to the clinical and demographic characteristics of the patients. The agreed-upon score was adopted for the final data set. In cases of disagreement in scoring, the process was repeated, and the final score was determined by consensus of both observers.
An eyepiece net with a regular array of 100 square lattices was used. Two methods were used to evaluate positive signals for the studied molecules in tubulointerstitum according to cellular staining patterns. For clear-cut individual cell staining including tubular cell nuclei staining for activated NF-κB or AP-1 by SWH and their subunits (p65/p50, c-jun/c-fos) by IHC, as well as interstitial cell staining for various biomarkers, the number of positively staining cells was determined in a sequence of 10–70 consecutive visual fields of ×40 high-power fields (0.0625 mm2 per square lattice) per renal biopsy. The only field adjustments were made to avoid glomeruli and large vessels. Results were expressed as the mean number of positive cells/mm2. For contiguous tubular cytoplasmic staining for IKK-α, CD40, CD40L, ICAM-1, IL-1β, TNF-a, IL-6, and GM-CSF, staining areas were evaluated by a seven-point scoring system as described previously by Rui-Mei et al. (1998). Briefly, the intensity of staining was graded as follows: 0 = absent staining, 1 = mild staining, 2 = moderate staining, and 3 = marked staining. Extent of staining was determined as follows: 0 (nil), 1 (<25%), 2 (25–50%), 3 (50–75%), and 4 (>75% of tubules stained). The grades were added together to obtain the final staining scores, ranging from 0 (negative staining) to 7 (maximal staining; marked intensity and >75% of the tubules stained).
Tubulointerstitial lesion (TIL) score was assessed as the measure of the severity of chronic tubulointerstitial injury including tubular lesions (TL) and interstitial fibrosis (IF) (Yamamoto et al. 1993). Morphological variables were graded on a semiquantitative scale of 0 to 3+ as follows: 0 (nil), 1+ (<25%), 2+ (25–50%), and 3+ (>50% of tubules) (Daniel et al. 2000). A total TIL score (0–6) was computed by summing the scores of each component.
Statistical Methods
Statistical analyses were performed using SigmaStat 3.1 programs (SPSS Inc.; Chicago, IL). Clinical and histopathological data (Table 1) were expressed as the mean ± SD. Results of semiquantitative data were expressed as the mean ± SE. Statistical significance was determined by one-way ANOVA followed by Bonferroni's test for multiple comparisons among various groups. For non-parametric data, one-way ANOVA on ranks followed by Dunn's test was used for multiple comparison testing. Correlation analyses were performed by calculating Pearson's correlation coefficients for parametric data and Spearman's correlation coefficients for non-parametric data; p values <0.05 were considered significant.
Results
Tubular Profiles
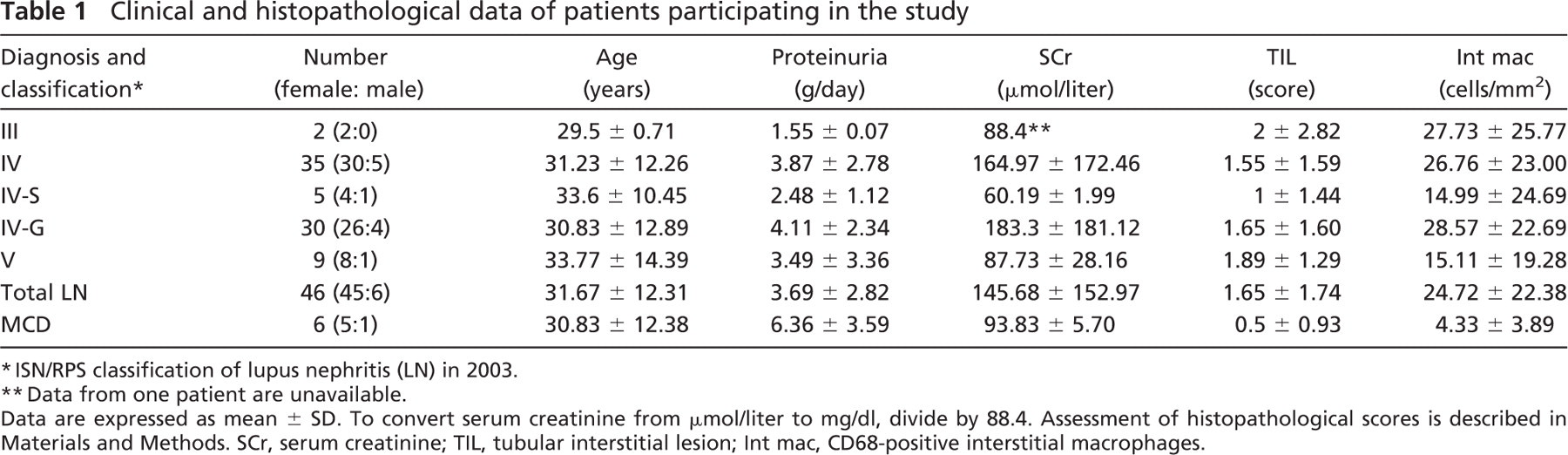
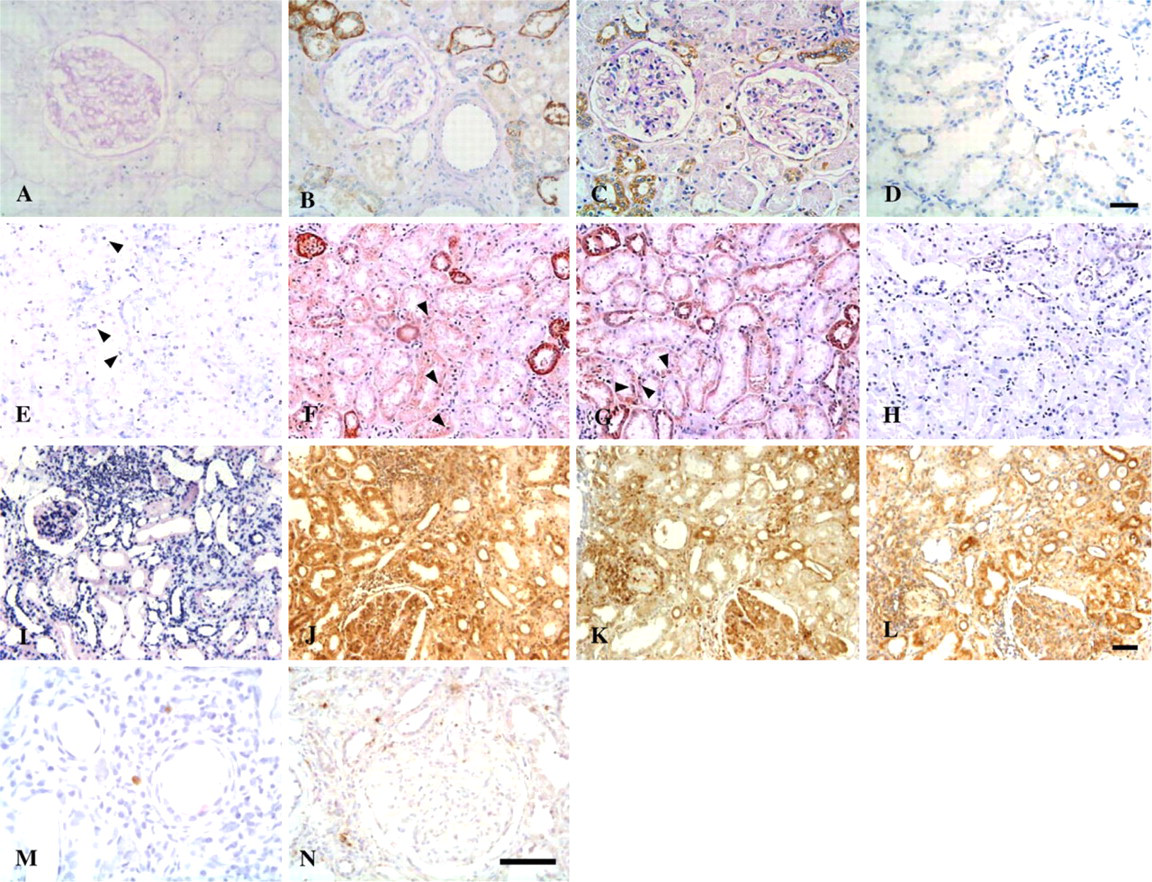
NF-κB Overactivation Concurrent With Upregulation of IKK-α in Tubular Epithelial Cells of Human LN. Tubular epithelial cell (TEC) nuclear staining for NF-κB by SWH was sparsely detected in normal kidneys (Figure 1A). Consistent with SWH results, expression of NF-κB subunits p65 and p50 was barely detectable in nuclei of TECs but was readily detectable in the cytoplasm in normal control samples (Figures 1B and 1C). In contrast, enhanced nuclear and cytoplasmic staining for NF-κB, p65, and p50 was noted in both intact and damaged cortical tubules in a patient with LN (Figures 1I–1K). Tubular expression levels of p65 and p50 were positively correlated in LN samples (Pearson's coefficient r = 0.57, p<0.001). Although tubular expression of activated NF-κB, p65, and p50 was also upregulated in MCD as compared with normal controls (Figures 1E–1G), the average number of TEC nuclei positive for NF-κB and p50 in MCD was significantly less than that observed in LN (Figure 2A).
Normal kidneys and MCD both demonstrated faint immunoreactivity for IKK-α in renal TECs (Figures 1D and 1H). In contrast, pronounced upregulation of IKK-α (Figure 1L) was observed in renal TECs from the same LN sample shown previously in Figures 1I–1K, as confirmed by semiquantitative analysis (Figure 2B). Notably, expression of IκB-α and p-IκB-α was undetectable in cortical tubules in various comparison groups including LN (Figures 1M and 1N).
Enhanced Parallel Expression of Activated AP-1 With NF-κB in Human LN. In normal controls, nuclear staining of AP-1 and its subunits c-jun and c-fos was occasionally detected in some TECs (Figures 3A–3C). Nuclear staining of AP-1 and its subunits c-jun and c-fos was prominent in renal TECs from the same LN sample shown previously in Figures 1I–1L, in a pattern similar to that of activated NF-κB (Figures 3G–3I). Increased tubular expression of AP-1 and its subunits was also found in MCD (Figures 3D–3F) but was significantly lower than in LN (Figure 2A). Moreover, tubular activation of NF-κB and AP-1 was strongly correlated in LN patients (Pearson's coefficient r = 0.732, p<0.001), whereas no significant correlation was found in the control MCD patients (Spearman's coefficient r = 0.429, p = 0.397).
Enhanced Tubular Expression of Signaling Molecules CD40/CD40L and NF-κB-regulated Molecules (TNF-α, IL-1β, ICAM-1, IL-6, GM-CSF) in Human LN. Normal controls and MCD patients showed absent or faint immunoreactivity for CD40 and CD40L in distal tubules (Figures 4A, 4B, 4E, and 4F). Increased expression of CD40 and CD40L was mainly localized in damaged renal tubules in LN as compared with normal controls and MCD (Figures 4I and 4J and Figure 5). Tubular expression of NF-κB-regulated molecules ICAM-1, IL-1β, IL-6, and GM-CSF was enhanced and most pronounced in damaged tubules of LN patients (Figures 4D, 4H, and 4L and Figure 6) as compared with normal controls and patients with MCD (Figure 5). In contrast to sparse staining of TNF-α in normal controls (Figure 4C), TNF-α expression was pronounced in both intact and damaged tubules of LN patients (Figure 4K) but did not differ from that of MCD patients (Figure 4G and Figure 5). Immunostaining of ICAM-1 and TNF-α was localized predominantly on the apical surface of cortical tubules, whereas CD40 was expressed mainly along the basal membrane of tubules, and tubular expression of other molecules was predominantly observed to be cytoplasmic or a mixed pattern (Figure 4 and Figure 6).
In situ detection of activated NF-κB, IKK-α, IκB-α, and p-IκB-α in renal tissues. (
Interstitial Cells
We observed increased numbers of interstitial cells that were immunopositive for activated transcription factors (NF-κB and AP-1) and their corresponding subunits in LN as compared with MCD and normal controls (Figure 1 and Figure 3; Table 2). IKK-α was concomitantly expressed with NF-κB in interstitial infiltrates in patients with LN (Figure 1L; Table 2). In contrast to their absence in tubular cells, IκB-α and p-IκB-α staining was readily detected in interstitial cells (Figures 1M and 1N; Table 2).
Semiquantitative analysis of tubular expression of activated transcription factors (NF-κB and AP-1), IKK-α, and cytokines among comparison groups. (
CD40- and CD40L-positive interstitial infiltrates were frequently seen in patients with LN (9.35 ± 2.46 and 15.83 ± 5.30, respectively) (Figures 4I and 4J). Tubular expression of CD40 positively correlated with the number of interstitial cells that stained immunopositive for CD40L (Spearman's coefficient r = 0.63, p=0.001). Additionally, there were increased numbers of interstitial cells positive for ICAM-1 (37.98 ± 5.39), TNF-± (6.64 ± 1.49), IL-1β (28.50 ± 4.96), IL-6 (7.85 ± 2.33), and GM-CSF (14.19 ± 3.38) in LN patients in comparison with MCD patients (ICAM-1: 0.51 ± 0.32; other markers: 0 ± 0) and normal controls (IL-1β: 3.63 ± 1.02; ICAM-1: 0.19 ± 0.16; other markers: 0 ± 0) (Figure 4 and Figure 6).
Correlation Analyses
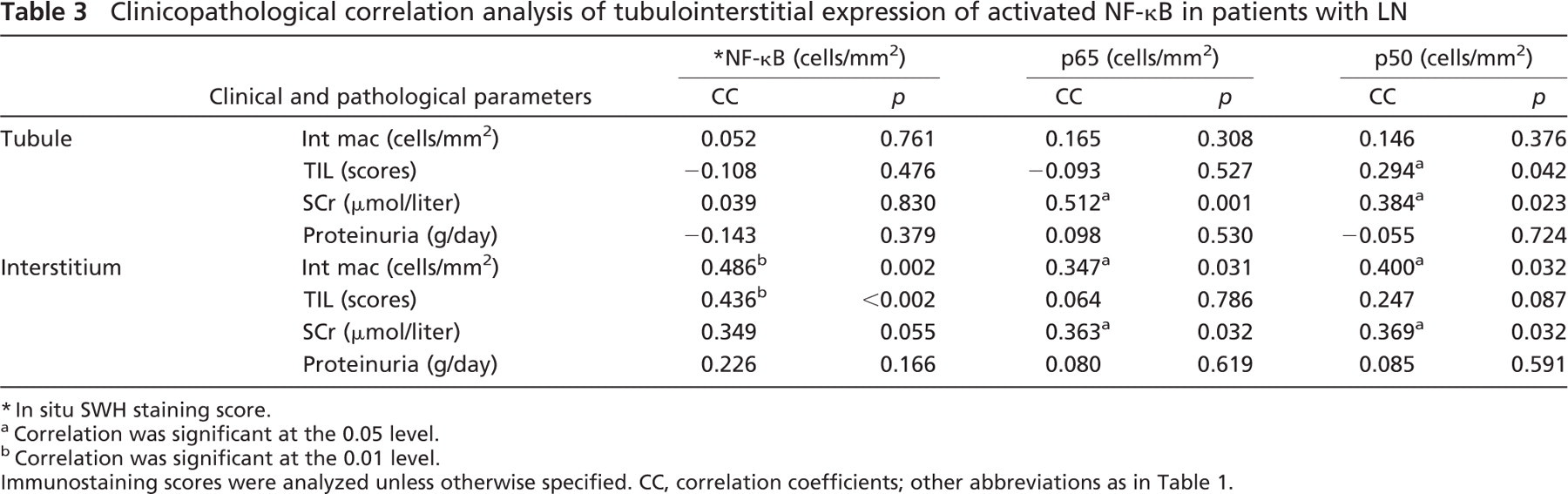
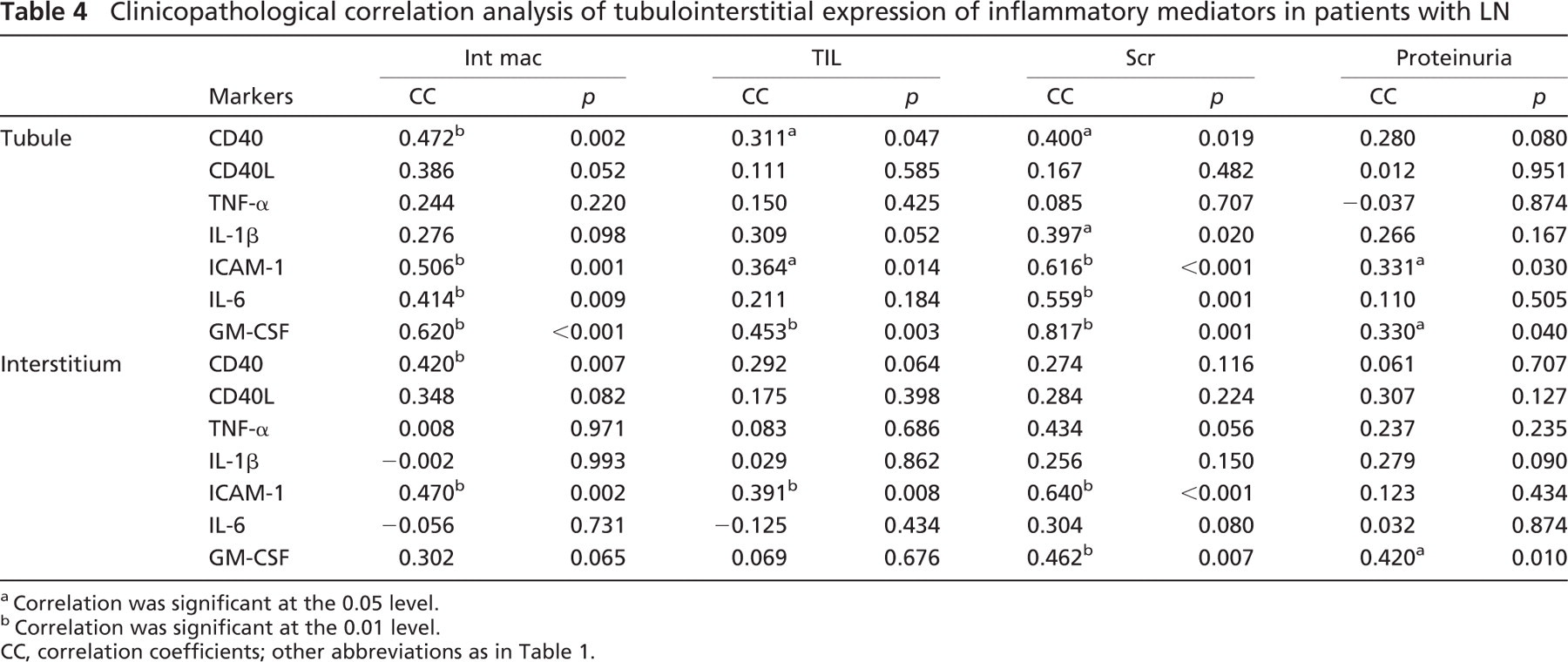
Clinicopathological Correlation Analysis. The average number of tubular cell nuclei that stained positively for p50 was weakly correlated with TIL scores, whereas a positive correlation was observed between tubular activation of p65 and p50 and the serum creatinine level (Table 3). TIL scores and/or the serum creatinine level were also positively correlated with tubular expression of CD40, IL-1β, ICAM-1, and GM-CSF with statistical significance or reaching marginal significance (Table 4). Positive correlations between tubular expression of CD40, ICAM-1, IL-6, and GM-CSF and interstitial infiltration of macrophages were observed (Table 4). There was a weak correlation between tubular activation of ICAM-1 and GM-CSF and the magnitude of proteinuria (Table 4). No significant correlations were observed between tubular expression of AP-1 or its subunits (c-jun and c-fos) and clinicopathological indices (data not shown).
Interstitial expression of activated NF-κB, p65, and p50 positively correlated with histopathological indices and serum creatinine level (Table 3), whereas interstitial expression of AP-1 positively correlated with interstitial infiltration of macrophages (Spearman's coefficient r = 0.419, p=0.030). Moreover, interstitial expression of CD40 positively correlated with tubulointerstitial histological indices, whereas interstitial expression of ICAM-1 was positively correlated with both histological indices and serum creatinine level (Table 4). Interstitial expression of GM-CSF positively correlated with proteinuria and serum creatinine level; a trend toward a positive correlation with interstitial infiltration of macrophages was also observed, which nearly reached statistical significance (Table 4). A trend toward a positive correlation between the number of interstitial cells expressing TNF-α and the level of serum creatinine was also observed, which did not reach statistical significance (Table 4).
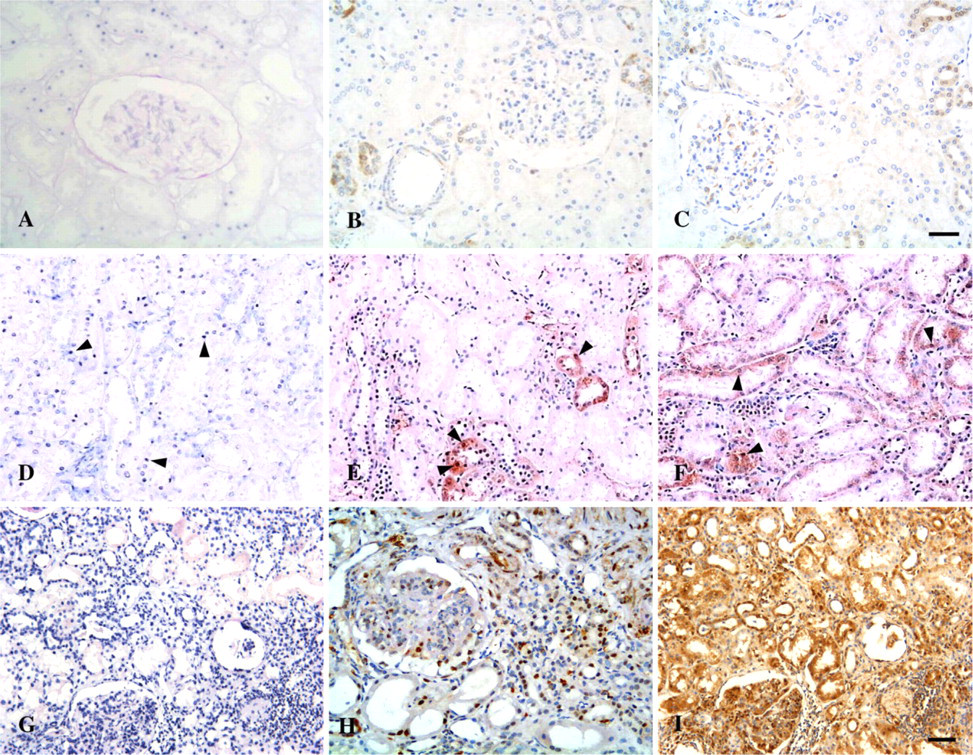
Microphotographs of tubulointerstitial expression of activated AP-1 in patients with LN and normal controls. (
Correlation Analysis Between Tubulointerstitial Expression of Activated NF-κB and IKK-α LN. We observed positive correlations between tubulor expression of IKK-α and both activated NF-κB and p50 (Spearman's coefficient r = 0.384, p=0.033; Spearman's coefficient r = 0.359, p=0.047), whereas marginal correlations were observed between interstitial expression of NF-κB and IKK-α (Pearson's coefficient r = 0.360, p=0.050).
Discussion
The major novel finding of our study is that activated transcription factor NF-κB is extensively upregulated in the renal tubular cells of LN patients as compared with normal controls and MCD patients. Expression pattern of p65 and p50 was similar to that of NF-κB detected by SWH in the tubulointerstitium of the LN group. A significant statistical association was found between nuclear immunopositive staining in tubular cells for p50 and p65, which comprises the two components of the “classic” NF-κB heterodimer. In addition to the “classic” NF-κB heterodimer, other subunits such as c-rel, p52, or RelB have also been reported to participate in transcriptional upregulation of inflammatory mediators in renal tubular cells (Morrissey and Klahr 1997). Expression of multiple distinct forms of NF-κB heterodimers is consistent with our finding that the number of SWH-stained tubular cell nuclei (overexpressing activated NF-κB) is overwhelmingly higher than that of p65- or p50-stained tubular cell nuclei in LN samples. Tubular activation of p65 and p50 positively correlated with the degree of renal function and/or tubulointerstitial histopathological indices, indicating a pathogenic role for the “classic form” of NF-κB in the progression of tubulointerstitial lesions. Notably, such a clinicopathological correlation was not seen for SWH-stained tubular cell nuclei. This finding is likely attributable to the lesser or indirect role of “non-classic” forms of NF-κB in the progression of tubulointerstitial lesions in LN samples.
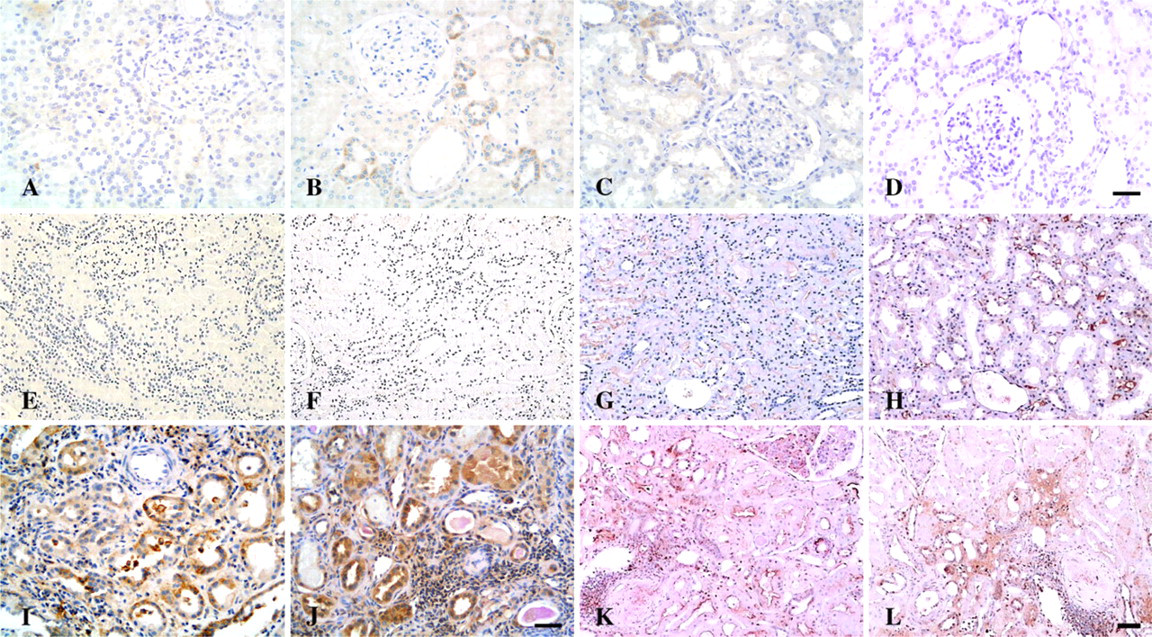
Microphotographs of immunostaining for CD40, CD40L, TNF-α, and IL-1β in controls and patients with LN. (
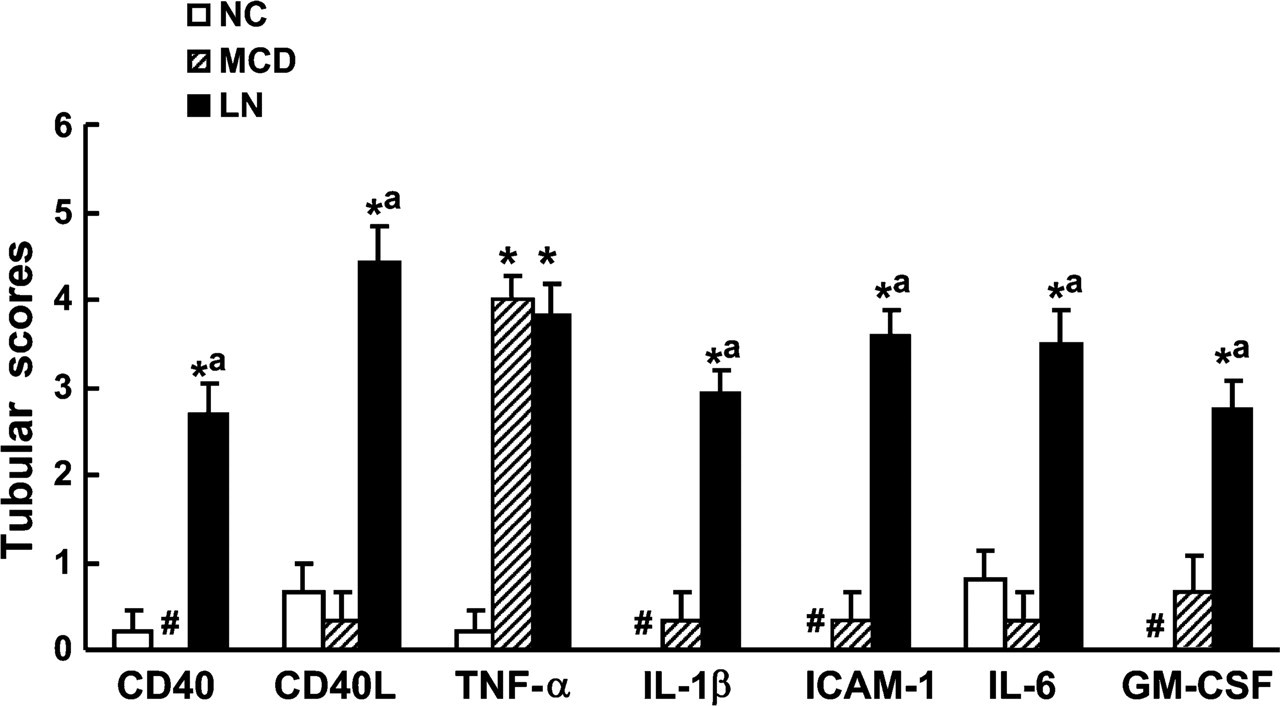
Semiquantitative analysis of tubular expression CD40, CD40L, TNF-α, IL-1β, ICAM-1, IL-6, and GM-CSF. ∗p<0.05 vs NC; a p<0.05 vs MCD; #no marker expression is detectable. NC, normal control; MCD, minimal change disease; LN, lupus nephritis.
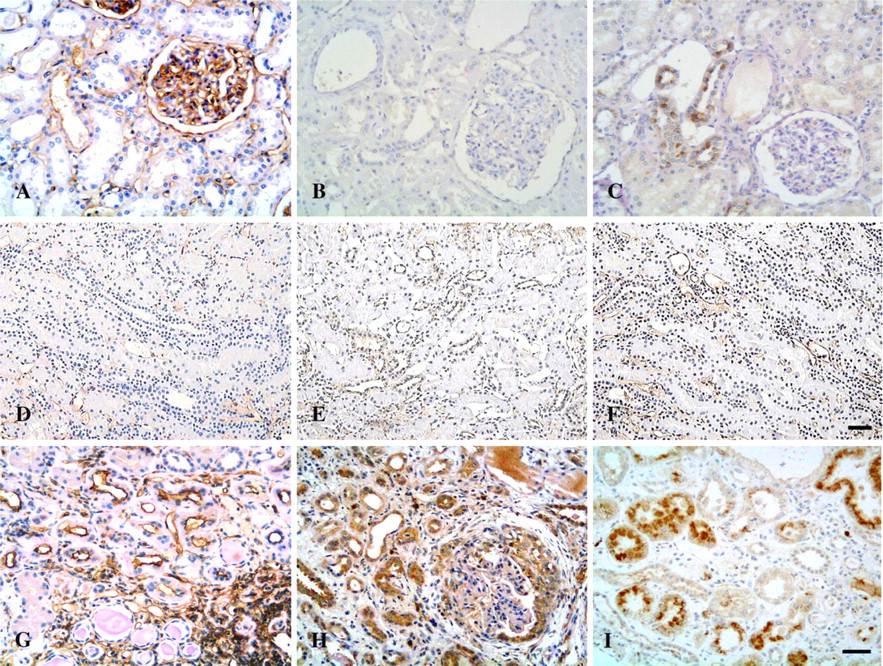
Microphotographs of tubulointerstitial expression of ICAM-1, GM-CSF and IL-6 in controls and patients with LN. (
In the present study, tubular overexpression of NF-κB downstream inflammatory mediators ICAM-1, IL-1β, IL-6, and GM-CSF was found to be positively correlated with the degree of renal function and/or tubulointerstitial histolopathological indices in LN samples, consistent with our proposal that NF-κB is a key mediator of tubulointerstitial injury. The proinflammatory role of NF-κB activation in renal tubular cells has been implicated in tubulointerstitial injury in the proteinuria-induced rat model through transcriptional activation of NF-κB-dependent inflammatory mediators (Rangan et al. 1999; Gomez-Garre et al. 2001; Takase et al. 2003). NF-κB augments expression of adhesion molecules such as ICAM-1 and VCAM-1 in renal tubular cells (Oertli et al. 1998; Tu et al. 2001). Proinflammatory cytokines such as TNF-α, IL-1, IL-6, and GM-CSF are also regulated by NF-κB in tubular epithelial cells or glomerular epithelial cells (Guijarro and Egido 2001; de Haij et al. 2002; Drumm et al. 2002; Greiber et al. 2002; Viedt et al. 2002). These inflammatory molecules have been implicated as critical mediators in the progression of tubulointerstitial lesions (Healy and Brady 1998). However, associations between NF-κB/TNF-α/IL-6 and tubulointerstitial lesion scores were not observed in this study. This is likely due to the existence of pleiotrophic pathways mediated by these molecules in renal inflammation. In our previous report, associations between NF-κB and proliferation in renal tubular cells suggested a pro-proliferative role for NF-κB in tubulointerstitial inflammation (Zheng et al. 2006b). An anti-apoptotic role for TNF-α has been reported in opossum kidney cells, proposed to be mediated by actin redistribution that involves NF-κB activation (Papakonstanti and Stournaras 2004). An anti-inflammatory role of IL-6 has also been suggested, apart from a pro-inflammatory role (Xing et al. 1998).
Interstitial expression of NF-κB, AP-1, their subunits, and NF-κB modulators in patients with LN
In patients with MCD, only tubular overexpression of TNF-α was associated with NF-κB activation in tubulointerstitial regions exhibiting minor degrees of injury. The role of TNF-α and NF-κβ activation produced by tubular cells in patients with MCD remains unknown. Cellular response may be a secondary response to albuminuria itself, rather than playing a causative role in the pathogenesis of MCD (Cho et al. 2003).
Clinicopathological correlation analysis of tubulointerstitial expression of activated NF-κB in patients with LN
Cooperation among NF-κB and other transcription factors such as AP-1 is required for effective induction of ICAM-1, GM-CSF, TNF-α, and IL-6 (Tsuboi et al. 1994; Dendorfer 1996; Sakiri et al. 1998; Viedt et al. 2002; Blaber et al. 2003). Cross-coupling between NF-κB and AP-1 resulting in a synergistic increase in activity at both AP-1 and NF-κB consensus DNA binding sites has been previously described (Adcock 1997). In experimental and human GN, AP-1 activation together with NF-κB overactivation has been found in tubular cells (Mezzano et al. 2001; Ruiz-Ortega et al. 2001). In the present study, tubular activation of AP-1 and its subunits in LN patients also exceeded that of normal control and MCD patients and significantly correlated with tubular NF-κB activation in LN. Results suggest that AP-1 activation may contribute to tubulointerstitial injury in cooperation with NF-κB in LN and that the differential expression of activated AP-1 in renal tubules between the LN and MCD groups may account, at least in part, for the observed difference in tubular expression of inflammatory mediators between groups.
We investigated the possible regulatory mechanisms of NF-κB activation in the kidney tissue of LN patients. IκB and IKK have been suggested to be involved in the regulation of NF-κB activation in renal tubular cells in vitro (Wang et al. 2000; Tu et al. 2001). In the present study, IKK-α expression was augmented in tubular cells in parallel with tubular activation of NF-κB in LN samples, suggesting the presence of an IKK–NF-κB pathway in renal tubular cells of LN. It has been proposed that receptor-mediated signaling by molecules such as CD40L and TNF-α may trigger activation of IKK, leading to phosphorylation of IκB, which then dissociates from NF-κB and is rapidly degraded, resulting in translocation of unbound NF-κB to nuclei (Hsing et al. 1997; Tu et al. 2001). Induction of NF-κB activation and downstream target genes by CD40/CD40L signaling has been documented in renal tubular cells (Woltman et al. 2000). In addition to TNF-α, elevated expression of CD40 and CD40L was observed in tubular cells and interstitial leukocytes and was associated with the severity of tubulointerstitial injury in LN in the current study. It is likely that IKK-α mediates CD40/CD40L signaling-induced NF-κB activation.
Clinicopathological correlation analysis of tubulointerstitial expression of inflammatory mediators in patients with LN
We were unable to detect expression of IκB-α and phosphorylated IκB-α (at Ser32) in renal tubules of the various comparison groups in the present study. Whether this is attributable to poor sensitivity of the monoclonal antibodies used or to technical factors remains unclear. It is possible that IκB-α, which is known to be degraded rapidly, is expressed at a level beyond the limit of sensitivity for the immunohistochemical detection methods used in the present study. Alternatively, other isoforms of IκB such as IκB-β may play an essential role in NF-κB activation in renal tubular cells. Recent studies indicate that the majority of p50/p65 complexes are regulated not only by IκB-α but also by IκB-β (Ghosh et al. 1998). IκB-α is thought to maintain the transient effect of inducing agents on the transcription of NF-κB responsive genes, whereas persistent activation of NF-κB is regulated by IκB-β in the nucleus (Ghosh et al. 1998). Thus, it is possible that persistent NF-κB activation in renal tubular cells is dependent on the IκB-β pathway in LN. This hypothesis deserves further investigation.
NF-κB and AP-1 activation also correlated well with interstitial infiltration of macrophages in LN samples. Recent studies suggest that NF-κB signaling is involved in activation of macrophages (Kluth et al. 2004). We observed IKK-α expression in interstitial cells in parallel with NF-κB activation in LN samples. Expression of IκB-α and, to a lesser extent, phosphorylated IκB-α was also detected in interstitial cells in LN, suggesting that the IKK-induced IκB-α phosphorylation pathway may contribute to NF-κB activation in interstitial cells. Interstitial expression of ICAM-1, CD40/CD40L molecules, and proinflammatory cytokines and their positive correlation with the severity of tubulointerstitial injury and/or renal function further support the hypothesis that renal tubular cells and interstitial infiltrates such as macrophages are involved in an interplay through a network of inflammatory mediators, which is crucial for the progression of tubulointerstitial injury in LN (Kuroiwa et al. 2000).
In conclusion, results of our study suggest that complex interactions between tubular cells and interstitial leukocytes through a network of inflammatory mediators may be integrated at the level of gene activation by the upregulation of transcription factors such as NF-κB and AP-1 in human LN. The IKK-induced NF-κB activation pathway is likely to be involved in both initiation and maintenance of tubulointerstitial injury by promoting production of proinflammatory mediators.
Footnotes
Acknowledgements
This work was funded by the Academic Research Fund Grant R-172-000-109-112 (to RS and SI-HH) and intramural funding from the Department of Medicine, National University of Singapore.
We thank Susan Lisa Nasr for assistance with the preparation of this manuscript. This work was undertaken in the Special Histopathology Laboratory in the Department of Pathology. We thank Dr. Koh Dow Rhoon for valuable advice and Dr. Thian Chai Lee (Johor Specialist Center, Malaysia) for assistance in providing the clinical data on the biopsy cases included in the present study.