
Abstract
Targeted proteomics research, based on the enrichment of disease-relevant proteins from isolated cell populations selected from high-quality tissue specimens, offers great potential for the identification of diagnostic, prognostic, and predictive biological markers for use in the clinical setting and during preclinical testing and clinical trials, as well as for the discovery and validation of new protein drug targets. Formalin-fixed and paraffin-embedded (FFPE) tissue collections, with attached clinical and outcome information, are invaluable resources for conducting retrospective protein biomarker investigations and performing translational studies of cancer and other diseases. Combined capillary isoelectric focusing/nano-reversed-phase liquid chromatography separations equipped with nano-electrospray ionization-tandem mass spectrometry are employed for the studies of proteins extracted from microdissected FFPE glioblastoma tissues using a heat-induced antigen retrieval (AR) technique. A total of 14,478 distinct peptides are identified, leading to the identification of 2733 non-redundant SwissProt protein entries. Eighty-three percent of identified FFPE tissue proteins overlap with those obtained from the pellet fraction of fresh-frozen tissue of the same patient. This large degree of protein overlapping is attributed to the application of detergent-based protein extraction in both the cell pellet preparation protocol and the AR technique.
B
The ability to identify proteins within FFPE tissue specimens is greatly enhanced by a simple and effective antigen retrieval (AR) technology, in which boiling the FFPE tissue sections in water or buffer solution dramatically reduces the detection thresholds (increases sensitivity) of immunohistochemistry (IHC) staining for a wide range of antibodies (Shi et al. 1991, 1997, 2001). In this context, IHC as applied to the demonstration of antigens (primarily proteins) in tissue sections represents a first form of protein analysis. The Human Protein Atlas compiles results from the systematic application of IHC to a wide variety of human tissues across many disease states (Nilsson et al. 2005; Uhlen and Ponten 2005; Uhlen et al. 2005). The mechanism of AR appears to involve a re-naturation of the structure of fixed proteins through a series of conformational changes, including the possible breaking (hydrolysis) of formalin-induced cross-linkages, the entire process driven by thermal energy from the heat source (Shi et al. 1991; Gown 2004). Still, the high-temperature heating treatment may also induce negative results, such as additional protein modifications, and may therefore require further studies and comparisons with lowtemperature heating treatment and combined retrieval protocols involving heat and enzyme digestion (Shi et al. 2000).
On the basis of the principle of heat-induced AR retrieval technique, Ikeda et al. (1998) extracted proteins from FFPE tissues by heating the sections in a radioimmunoprecipitation buffer containing 2% sodium dodecyl sulfate (SDS). Subsequently, Yamashita and Okada (2005) performed protein extraction from FFPE tissues by autoclaving the sections, followed by incubating the sections with a high-pH solution containing urea, 2-mercaptoethanol, and 2% SDS. A commercial Liquid Tissue kit, apparently based on the same AR retrieval technique, was recently introduced for processing FFPE tissues also in the presence of detergent while heating at 95C for 90 min (Desiere et al. 2005; Prieto et al. 2005).
In addition to IHC-based tissue proteome studies, current protein/peptide separation platforms, including two-dimensional polyacrylamide gel electrophoresis (2-D PAGE) and multidimensional liquid chromatography systems (Washburn et al. 2001; Wolters et al. 2001), require large cellular samples to increase proteomic coverage. Thus, most tissue proteomic studies have employed multidimensional liquid chromatography separations (Li et al. 2004; Zhang et al. 2004; Cagney et al. 2005; DeSouza et al. 2005) and are mainly based on analysis of entire tissue sections instead of targeted subpopulations of microdissection-derived cells. However, the heterogeneous nature of most tissues, as well as the fact that in many cases, the cells of interest may be in the minority and may be surrounded by normal or other abnormal cells, limits the ultimate utility of whole-tissue proteome studies in many instances.
Several tissue microdissection technologies, including laser capture microdissection (Emmert-Buck et al. 1996; Bonner et al. 1997), laser catapulting (Schutze et al. 1998), laser microdissection (DeSouza et al. 2005), and laser-free microdissection (Furuta et al. 2004; Zhuang et al. 2004), have been developed to provide a rapid, straightforward method for procuring homogeneous subpopulations of cells or structures for biochemical and molecular biological analyses. However, the smaller quantities of samples available for micro-dissected populations have, to this point, restricted protein analyses to the use of only a single chromatography separation prior to tandem mass spectrometry (MS/MS) analysis and have limited the ability to mine deeper into the tissue proteome in recent studies (Wu et al. 2003; Zang et al. 2004; Baker et al. 2005; Crockett et al. 2005; Hood et al. 2005; Palmer-Toy et al. 2005). Because the sizes of human tissue biopsies are becoming significantly smaller due to the advent of minimally invasive methods and early detection and treatment of lesions, a more effective discovery-based proteome technology is critically needed to enable sensitive studies of protein profiles that will have diagnostic and therapeutic relevance.
The key to performing sensitive tissue proteome analysis, as demonstrated in our previous studies (Wang et al. 2005a, c), is to attain high-analyte concentrations in small peak volumes. We have employed electrokinetic focusing and the high resolving power of a capillary isoelectric focusing (Chen et al. 2003)-based multidimensional separation platform to enhance the dynamic range and detection sensitivity of mass spectrometry (MS) measurements. By coupling with the heat-induced AR technique, combined capillary isoelectric focusing (CIEF)/nano-reversed-phase liquid chromatography (nano-RPLC) separations have been demonstrated for the analysis of proteins extracted from entire FFPE tissue sections (Shi et al. 2006). Instead of using whole sections, minute proteins procured from microdissected glioblastoma multiforme (GBM) FFPE tissues, as the model proteome system employed in this study, are processed, profiled, and compared with those extracted from fresh-frozen tissues of the same, matched patient. The work reported here represents an important milestone toward the development, evaluation, and validation of a novel biomarker discovery paradigm based on years of archived FFPE tissue collections.
Materials and Methods
Clinical Materials
Tissues and clinical (pathological) information were obtained as part of an Institutional Review Board-approved study at the Cleveland Clinic. At the time of craniotomy, tissue samples were split equally; one portion was sent for routine processing in the pathology department, and the other was snap-frozen, as noted below. For the portion sent to pathology, from which a clinical diagnosis was made, the sample was processed in the routine fashion, fixed in formalin overnight, embedded in paraffin, and stored at room temperature after use. Approximately 6–12 months later, 6-μm-thick unstained sections were cut from this block and used for analysis, as described below. The second portion of tissue was immediately snap-frozen in liquid nitrogen in the operating room, embedded in the Optimal Cutting Temperature Medium (Tissue-Tek, Sakura, Finetek; Torrence, CA), and stored at −80C.
Materials and Reagents
Fused-silica capillaries (50 μm i.d./375 μm o.d. and 100 μm i.d./375 μm o.d.) were acquired from Polymicro Technologies (Phoenix, AZ). Acetic acid, ammonium acetate, ammonium hydroxide, ampholyte 3–10, dithiothreitol (DTT), formic acid, iodoacetamide (IAM), and octane were obtained from Sigma (St. Louis, MO). Acetonitrile, hydroxypropyl cellulose (average molecular mass 100,000), SDS, tris(hydroxymethyl)-aminomethane (Tris), and urea were purchased from Fisher Scientific (Pittsburgh, PA). Sequencing-grade trypsin was obtained from Promega (Madison, WI). All solutions were prepared using water purified by a Nanopure II system (Dubuque, IA) and further filtered with a 0.22-μm membrane (Millipore; Billerica, MA).
Tissue Microdissection and Protein Sample Preparation
FFPE tissues were deparaffinized using octane, followed by vortexing and centrifugation. Both deparaffinized FFPE and fresh-frozen tissues were microdissected by following the procedures described in our previous studies (Zhuang et al. 1995; Furuta et al. 2004; Zhuang et al. 2004) In brief, from all tumor samples, every third consecutive slide was stained with hematoxylin and eosin for pathological evaluation. A semiquantitative cell count was performed on tumor-rich (>90% tumor cells) areas that were not compromised by inflammation, necrosis, or stromal or endothelial proliferation. Subsequently, these areas were subjected to selective tumor dissection from serial sections. Tumor dissection was performed manually on unstained sections under the microscope to avoid possible heating artifacts induced by laser-assisted technology or chemical artifacts induced by tissue staining (Gottlieb and Chavko 1987; Palmer-Toy et al. 2000). We collected ~100,000 cells from each specimen; these were obtained from one to ten consecutive sections, depending on the size and the tumor portion of the specimen on the slide. Procurement of normal brain or areas of inflammation, necrosis, hemorrhage, or stromal or vascular proliferation was strictly avoided.
The microdissected cells obtained from fresh-frozen tissues were placed directly into a microcentrifuge tube containing 8 M urea and 20 mM Tris-HCl at pH 8.0. The soluble proteins were collected in the supernatant by centrifugation at 20,000 × g for 30 min. Proteins in the supernatant were reduced and alkylated by sequentially adding DTT and IAM with final concentrations of 10 mg/ml and 20 mg/ml, respectively. The solution was incubated at 37C for 1 hr in the dark and then diluted 8-fold with 100 mM ammonium acetate at pH 8.0. Trypsin was added at a 1:40 (w/w) enzyme-to-substrate ratio, and the solution was incubated at 37C overnight. Tryptic digests were desalted using a Peptide MacroTrap column (Michrom Bioresources; Auburn, CA), lyophilized to dryness using a SpeedVac (Thermo; San Jose, CA), and then stored at −80C.
In addition to acquiring the soluble protein fraction of targeted GBM cells procured from fresh-frozen tissues, cell pellets were treated by a 1% SDS solution (Han et al. 2001; Wei et al. 2005) containing 20 mM Tris-HCl at pH 8.0, followed by centrifugation at 20,000 × g for 30 min. The supernatant containing the membrane protein fraction was placed in a dialysis cup (Pierce; Rockford, IL) and dialyzed overnight at 4C against 100 mM Tris-HCl at pH 8.2. The extracted and dialyzed proteins were denatured, reduced, alkylated, digested, desalted, and lyophilized using the same sample preparation protocol as applied to the soluble protein fraction described previously.
Similar to the procedures described in our previous studies (Shi et al. 2006), the microdissected GBM cells obtained from FFPE tissues were treated with a 20-mM Tris buffer (pH 9) containing 2% SDS, followed by heating at 100C on a heat block (VWR Scientific Products; West Chester, PA) for 20 min, then incubation at 60C in an incubator (Robbins Scientific; Sunnyvale, CA) for 2 hr. The soluble proteins were collected in the supernatant by centrifugation at 20,000 × g for 30 min. The supernatant was placed in a dialysis cup (Old et al. 2005) and dialyzed overnight at 4C against 100 mM Tris-HCl at pH 8.2. The extracted and dialyzed proteins were denatured, reduced, alkylated, digested, desalted, and lyophilized using the same sample preparation protocol as applied to the membrane protein fraction of fresh-frozen tissues described previously.
Integrated CIEF/Nano-RPLC Multidimensional Peptide Separations
On-line integration of CIEF with nano-RPLC as a multidimensional peptide and protein separation platform has been described in detail in previous work (Chen et al. 2003; Wang et al. 2005a, b, c) and was employed for systematically resolving peptide digests on the basis of their differences in isoelectric point and hydrophobicity. Briefly, an 80-cm-long CIEF capillary (100 μm i.d./365 μm o.d.) coated with hydroxypropyl cellulose was initially filled with a solution containing 2% ampholyte 3–10 and 1.5 mg/ml tryptic peptides. Peptide focusing was performed by applying an electric field strength of 300 V/cm and using solutions of 0.1 M acetic acid and 0.5% ammonium hydroxide as the anolyte and the catholyte, respectively. The current decreased continuously as the result of peptide focusing. Once the current reached ~10% of the original value, usually within 30 min, the focusing was considered to be complete.
Focused peptides were sequentially fractionated by hydro-dynamically loading into individual trap columns (3 cm × 200 μm i.d. × 365 μm o.d.) packed with 5-μm porous C18 reversed-phase particles. A constant electric field of 300 V/cm was applied across the CIEF capillary for maintaining analyte band focusing in the capillary throughout the loading procedure. Each peptide fraction was subsequently analyzed by nano-RPLC equipped with an Ultimate dual-quaternary pump (Dionex; Sunnyvale, CA) and a dual nano-flow splitter connected to two pulled-tip fused-silica capillaries (50 μm i.d. × 365 μm o.d.). These two 15-cm-long capillaries were packed with 3-μm Zorbax Stable Bond (Agilent; Palo Alto, CA) C18 particles.
Nano-RPLC separations were performed in parallel. A dual-quaternary pump delivered two identical 2-hr organic solvent gradients with an offset of 1 hr. Peptides were eluted at a flow rate of 200 nl/min using a 5–45% linear acetonitrile gradient (containing 0.02% formic acid) over 100 min, with the remaining 20 min for column regeneration and equilibration. Full scans were collected from 400–1400 m/z using a linear ion trap mass spectrometer (LTQ, Thermo-Finnigan; San Jose, CA), and five data-dependent MS/MS scans were gathered, with dynamic exclusion set to 18 sec. A moving stage housing two nano-RPLC columns was employed to provide electrical contacts for applying electrospray voltages and, most importantly, to position the columns in line with the orifice of the heated metal capillary in the nano-electrospray ionization (ESI) source at the start of each chromatography separation and data acquisition cycle.
Data Analysis
The Open Mass Spectrometry Search Algorithm (OMSSA) developed at the National Center for Biotechnology Information (Geer et al. 2004) was used to search the peak list files against a decoyed SwissProt human database. This decoyed database was constructed by reversing all 12,484 real sequences and appending them to the end of the sequence library. Searches were performed using the following parameters: 1.5-Da precursor ion mass tolerance, 0.4-Da fragment ion mass tolerance, 1 missed cleavage, alkylated Cys as a fixed modification and variable modifications of acetylated N terminus and Lys, and oxidated Met. Searches were run in parallel on a 12-node, 24-CPU Linux cluster (Linux Networx; Bluffdale, UT).
False-positive rates were determined using the method of Elias et al. (2005). Briefly, false-positive rates were calculated by multiplying the number of false-positive identifications (hits to the reversed sequences scoring below a given threshold) by 2 and dividing by the number of total identifications. Peptides identified below threshold, and also occurring as matches to the forward sequences, were not counted as false positives or true identifications. A curve was then generated by plotting E-value versus false-positive rate, and an E-value threshold corresponding to a 1% false-positive rate was used as the cutoff in this analysis (Rudnick et al. 2005). After generation of search data, the result files were parsed and loaded into a custom MySQL database for visualization and reporting using in-house software.
Results
In human disease research, where knowledge of disease outcome is critical for the evaluation of the significance of phenotypic or genotypic profiles, as well as response to therapy and outcome, it may take five, ten, or more years to gain a relatively complete picture of the pathophysiology of a disease. The ability to analyze well-characterized, archival cases is highly desirable. In addition, because the capacity to store large numbers of cataloged samples under optimal conditions is limited by cost, space, and personnel limitations, among others, the development of methods to analyze traditional pathological specimens, such as FFPE tissues, is an important priority. However, too often molecular analysis techniques are applied directly to these formalin-paraffin materials, or extracts thereof, without an understanding of the variables introduced by the effects of tissue fixation and processing, whether upon the structure or availability of DNA, RNA, and proteins. Accessibility of macromolecules in the fixed-tissue specimens is therefore a critical issue, exemplified by the growth of IHC for protein antigens, and in situ hybridization for DNA and RNA.
Technological innovations already allow RNA profiling of FFPE tissues for the studies of patterns of altered gene expression caused by specific exposures or disease outcomes (Lewis et al. 2001; Nyska et al. 2001). Formalin-induced cross-linking of proteins serving as an efficient means of in-situ preservation of proteins, however, greatly hinders efficient extraction of proteins from tissue sections and subsequent proteomic efforts. Building upon our initial success in the integration of the CIEF-based multidimensional separation platform with the AR technique (Shi et al. 2006), we have incorporated a selective, laser-free microdissection approach (Furuta et al. 2004; Zhuang et al. 2004) into the workflow of this study to enable comprehensive analysis of protein profiles within targeted tumor cells instead of whole FFPE tissue blocks.
As evaluated by SDS-PAGE (Figure 1), the quality of protein pattern within the soluble fraction of targeted tumor cells procured from fresh-frozen GBM tissue was superior to that extracted from microdissected FFPE tissue from the same patient. The smear among FFPE protein bands, particularly in the range of low-to-medium molecular masses, may be the result of protein fragmentation from the heat-induced AR process. Besides reinstating the condition of a formalin-modified protein back to its original structure, the high-temperature heating treatment has been reported to induce a variety of protein modifications, including fragmentation (Shi et al. 2000; Gown 2004).
In addition to the evaluation of intact proteins using SDS-PAGE, combined CIEF/nano-RPLC separations were employed for the examination of protein digests to provide further in-depth comparison of proteomes within microdissected fresh-frozen and FFPE GBM tissue specimens. For profiling tryptic peptides obtained from an FFPE tissue sample, the entire content of focused peptides in the CIEF capillary was split into 19 individual fractions (Figure 2), which were further resolved by nano-RPLC and identified using nano-ESI-LTQ-MS/MS. The number of distinct peptide identifications measured from each CIEF fraction is significantly greater than that typically reported in the literature using other IEF techniques, including immobilized pH gradient gels (Cargile et al. 2004; Essader et al. 2005) and gel-free approaches (Wall et al. 2000; Zuo et al. 2001; Yan et al. 2003; Moritz et al. 2004) such as chromatofocusing, immobilized pH membranes, Rotofor, and free-flow electrophoresis. A key feature of our CIEF-based multidimensional separation technology is the elimination of protein/peptide loss and dilution in an integrated platform while achieving comprehensive and ultrasensitive analysis of protein expression profiles within FFPE tissue specimens. By contrast, preparative-scale IEF techniques (Wall et al. 2000; Zuo et al. 2001; Yan et al. 2003; Cargile et al. 2004; Moritz et al. 2004; Essader et al. 2005) are incompatible with the smaller scale of the more selectively procured proteomes obtained from microdissection-procured tissues.
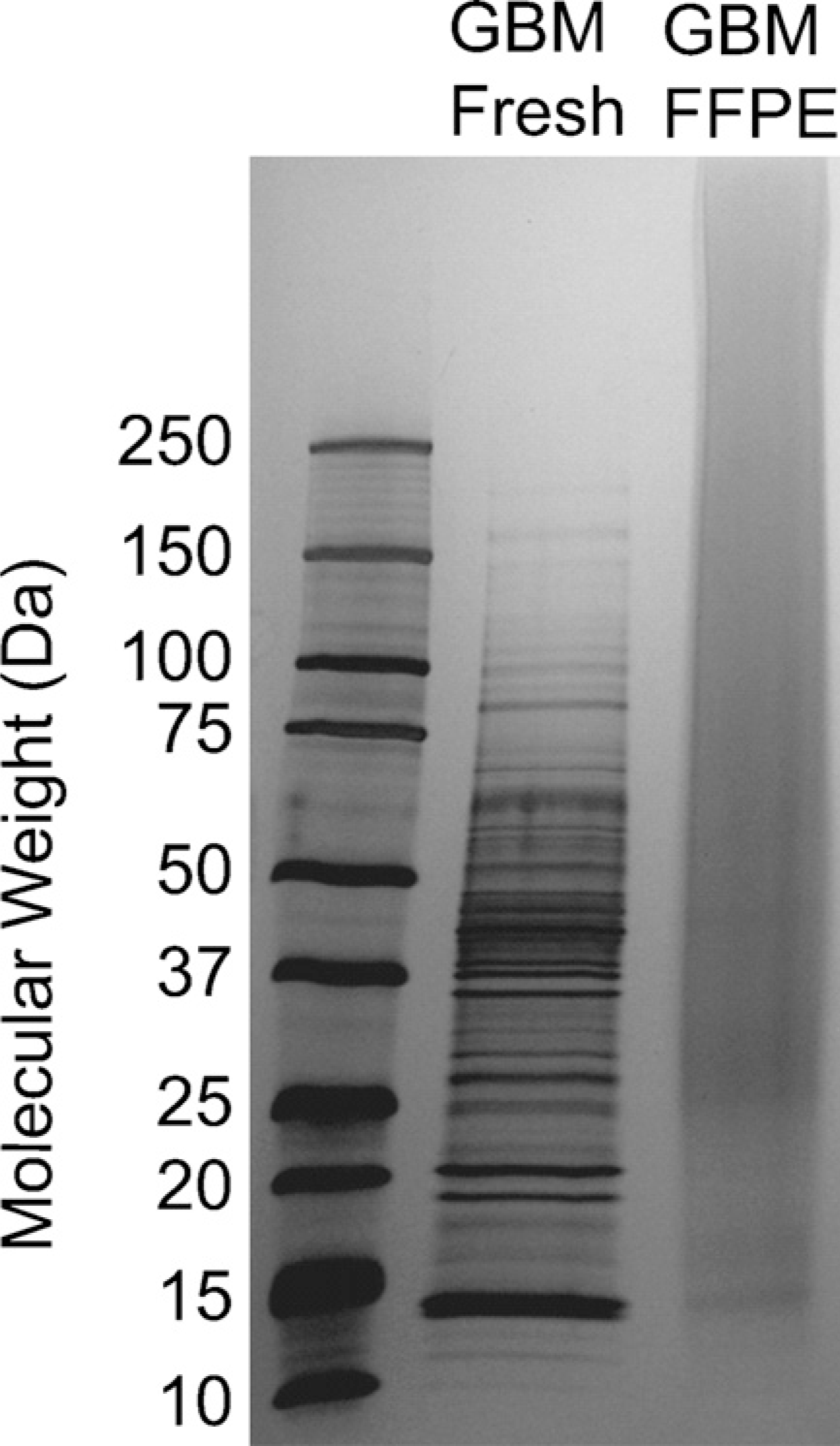
Comparison of protein profiles obtained from microdissected fresh-frozen (Lane 1) and formalin-fixed and paraffin-embedded (FFPE) (Lane 2) glioblastoma multiforme (GBM) tissue specimens from the same patient using SDS-PAGE.
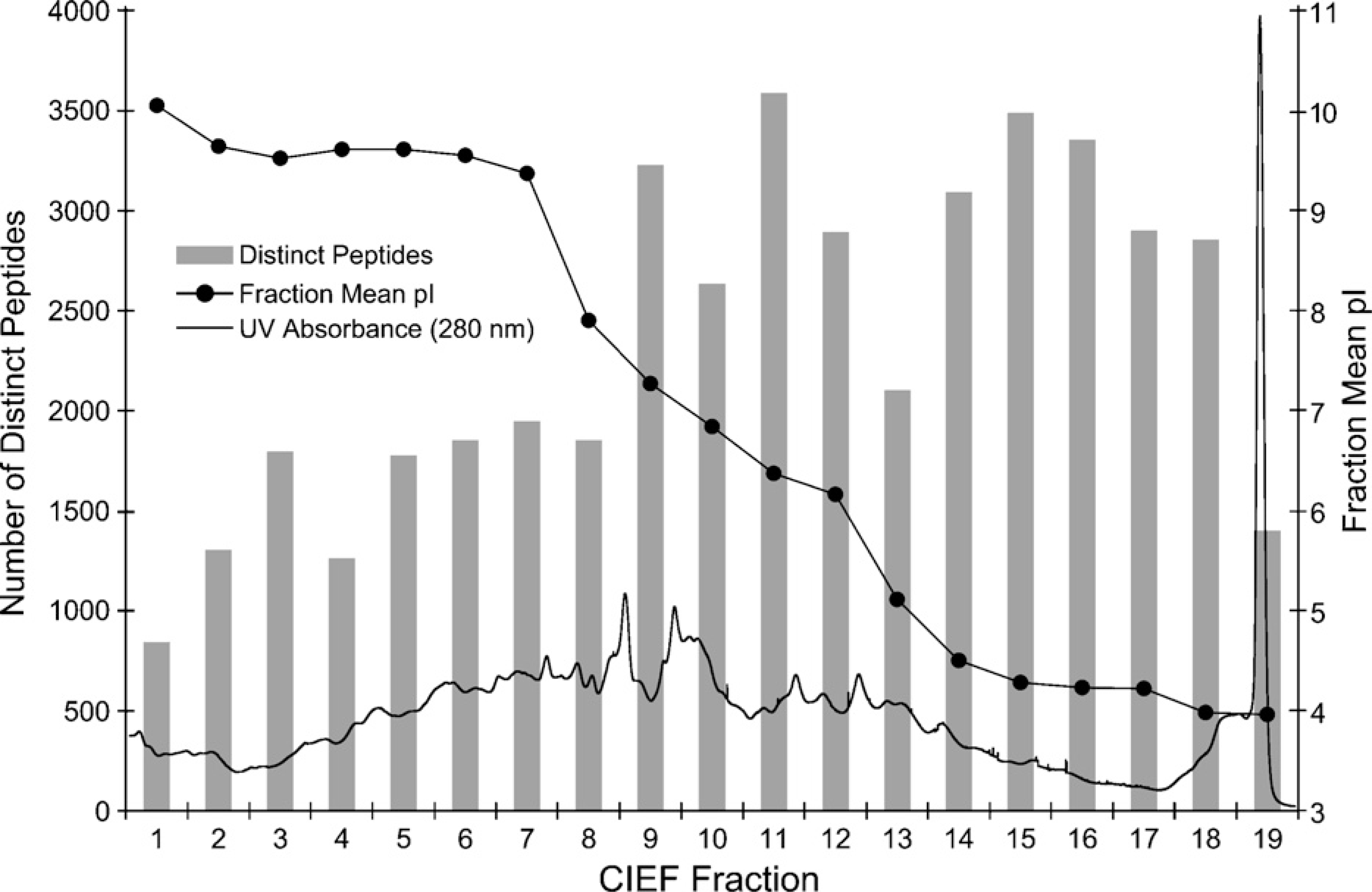
Overlaid plots containing the capillary isoelectric focusing (CIEF)-ultraviolet absorbance response trace monitored at 280 nm, the number of distinct peptides identified in each of the CIEF fractions, and the distribution of the peptides mean isoelectric point values over the entire CIEF separation.
As shown in Figure 3, the peptide and protein false-positive rates, and the numbers of total peptides, distinct peptides, and protein identifications were plotted as functions of the E-value of a typical OMSSA search. An E-value threshold of 0.17, corresponding to 1% false positive of total peptide identifications, was chosen as a cutoff for the FFPE sample. A total of 14,748 distinct peptides were identified, leading to the identification of 2733 non-redundant proteins from the SwissProt human database containing 12,484 non-redundant protein entries. This identity threshold also resulted in a protein false-positive rate of 7.5%, as indicated by the detection of peptides from 107 distinct reversed protein sequences in the decoy section of the search database.
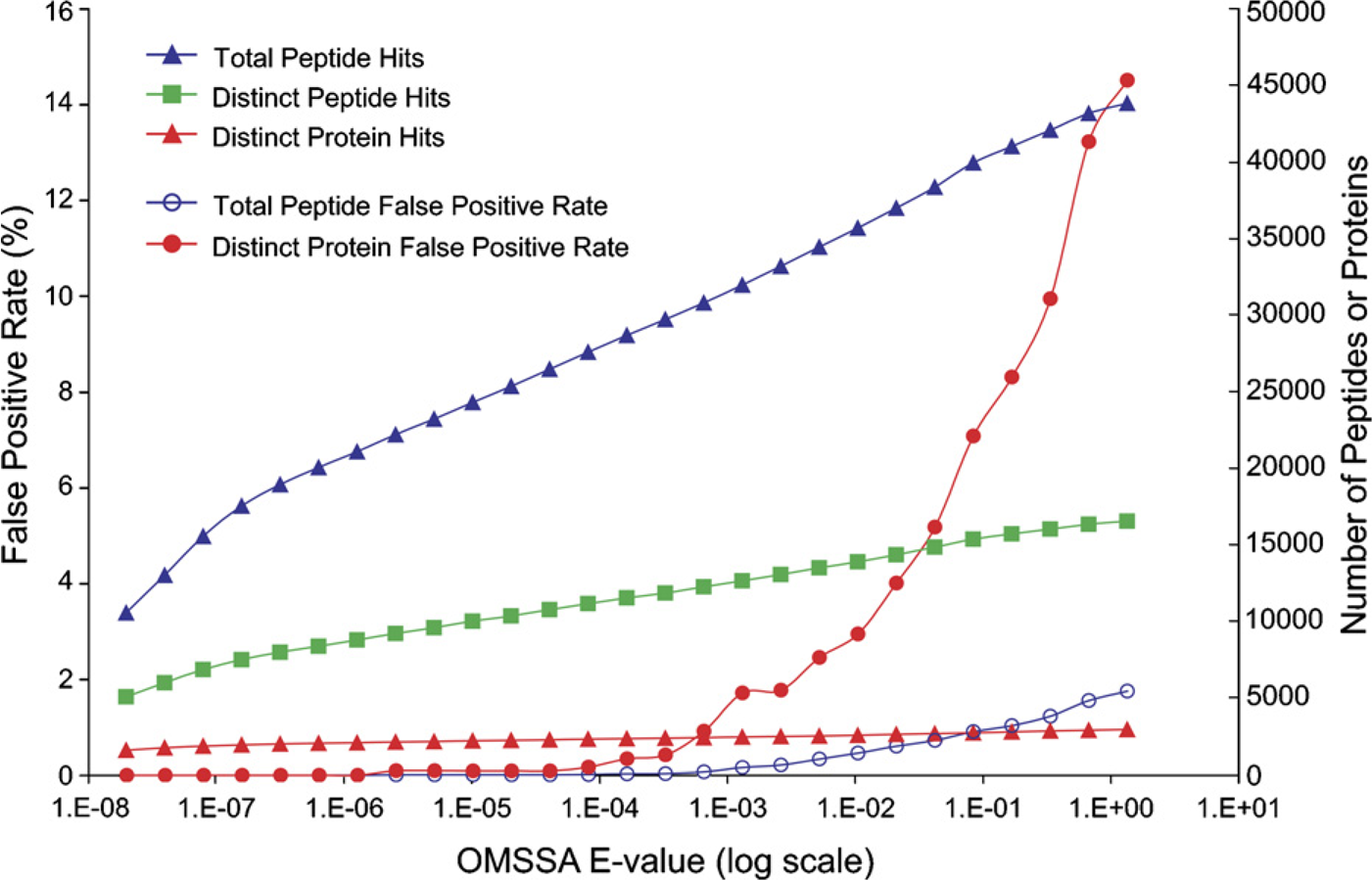
Plots of the false-positive rates and the numbers of total peptide, distinct peptide, and distinct protein identifications versus the E-value obtained from the search of the peak list files against a decoyed SwissProt human database using the Open Mass Spectrometry Search Algorithm (OMSSA).
The first reversed protein was detected at an E-value of 2 × 10−6. At this threshold score, a total of 10,755 distinct peptides were identified, leading to the identification of 2224 non-redundant proteins (Figure 3). By tolerating a 1% false positive of total peptide identifications (E-value threshold of 0.17), an additional 3993 distinct peptides and 509 distinct proteins were measured at a cost of 112 and 107 predicted false identifications of distinct peptides and proteins, respectively. By further increasing the E-value threshold to 1.3, the false-positive rates escalated to 1.8% and 15.0% for total peptide and protein identifications, respectively.
To better illustrate the impact of protein false-positive rate on protein identification, it should be emphasized that new distinct proteins were added to search results at a ratio of ~50:1 relative to reversed distinct proteins at an E-value of 2 × 10−6. At an E-value of 0.17, corresponding to a protein false-positive rate of 7.5%, this ratio decreased to 2:1. This ratio was further reduced to 1:1 at an E-value of 1.3, meaning that new forward protein sequences were added at a rate equal to that of reversed protein sequences. The implication is that all new forward protein sequences were probably false positives at an E-value of 1.3, which corresponded to the false-positive rates of 1.8% and 15.0% for total peptide and protein identifications, respectively.
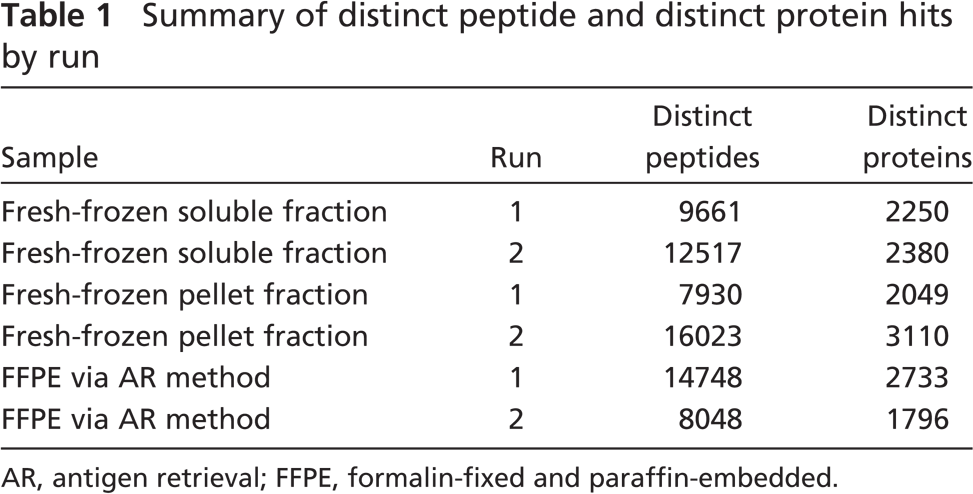
Compared with several recently reported FFPE tissue-based proteome studies (Crockett et al. 2005; Hood et al. 2005; Palmer-Toy et al. 2005), our results present the largest catalog of proteins from a single microdissected FFPE tissue specimen that we know of to date. Application of only a single-dimension separation in these recent studies (Crockett et al. 2005; Hood et al. 2005; Palmer-Toy et al. 2005) due to sample amount constraints has significantly limited the dynamic range and detection sensitivity of MS measurements, and greatly impacted their ability to mine deeper into the tissue proteome. A total of 2845 non-redundant proteins were identified from combined proteome runs of a single FFPE tissue sample. Table 1 presents a summary of the results of each run.
Summary of distinct peptide and distinct protein hits by run
AR, antigen retrieval; FFPE, formalin-fixed and paraffin-embedded.
Among proteins identified from the microdissection-procured FFPE GBM tissue specimen, 488 proteins were predicted to contain at least one or more transmembrane domains using TMHMM (www.cbs.dtu.dk/services/TMHMM-2.0/) (Krogh et al. 2001). The sub-cellular location of proteins identified from the FFPE tissue was assigned using the protein subcellular localization prediction tool (PSLT) (Scott et al. 2005). Still, the subcellular location of approximately half of the identified proteins cannot be assigned by PSLT. Among proteins with assigned locations, 34% were found in plasma membrane and organelle categories.
The sequence coverage of two representative transmembrane proteins, tenascin and basigin, is presented in Figure 4, together with the examples of peptides' tandem mass spectra leading to their identification. Tenascin, a glioma-associated extracellular matrix antigen, is a substrate adhesion molecule that appears to inhibit cell migration and may play a role in supporting the growth of epithelial tumors. Tenascin is also a ligand for integrins of α-8/β-1, α-9/β-1, α-V/β-3, and α-V/β-6 (Olson and Srivastava 1996). Basigin, a tumor cell-derived collagenase stimulatory factor, is enriched on the surface of tumor cells and upregulated in gliomas. Its expression level correlates with the malignant potential of the tumor (Spring et al. 1997). For its tissue specificity, basigin is only present in vascular endothelium in non-neoplastic regions of the brain, whereas it is present in tumor cells but not in proliferating blood vessels in malignant gliomas.
In addition to profiling the FFPE tissue proteome, combined CIEF/nano-RPLC separations coupled with nano-ESI-LTQ-MS/MS were employed in the analysis of protein digests obtained from the soluble and cell pellet fractions of microdissected fresh-frozen tissue. Fresh tissue taken from the same case of GBM was microdissected and processed for the extraction of soluble proteins using urea, followed by an SDS-based protocol (Han et al. 2001; Wei et al. 2005) for the preparation of membrane proteins from remaining cell pellets. By using a 1% false-positive rate for total peptide identifications, a total of 2856 and 3227 proteins were identified from the soluble and pellet fractions, respectively. By combining the proteome results obtained from the soluble and pellet fractions, the collective analysis yielded the identification of 3902 non-redundant proteins with an average of 6.2 peptides per protein overall, corresponding to 31% coverage of the SwissProt human database.
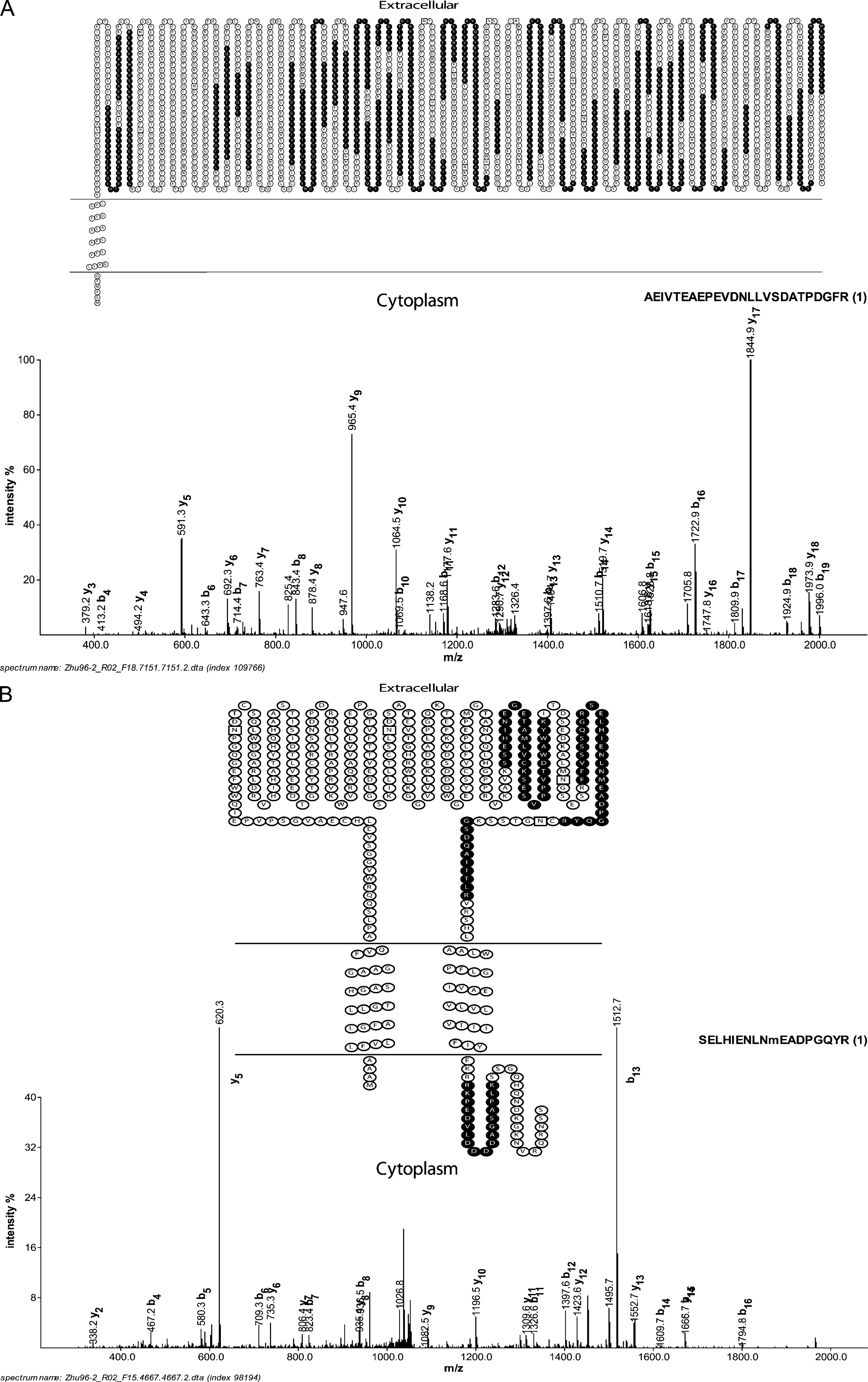
Peptide coverage of representative transmembrane proteins such as (A) tenasin and (B) basigin, and tandem MS spectra of unique peptides leading to their identification.
Comparing the proteome results obtained from the fresh-frozen and FFPE tissues (Figure 5), most proteins identified from the FFPE slide were also detected in the corresponding fresh-frozen section. Only 243 proteins, representing 8.5% of total protein identifications, were unique to the FFPE tissue. Among proteins identified from the FFPE tissue, 2370 proteins, or 83% of the total protein identifications, overlapped with those measured from the pellet fraction of fresh-frozen tissue. The percentage of overlap among membrane proteins (predicted to contain at least one or more transmembrane domains) identified from the FFPE and the pellet fraction of fresh-frozen tissues was ~83% and was further increased to 88% by including membrane proteins measured from the soluble fraction (Figure 6). We attribute our success with this high concordance in protein identification between FFPE and fresh-frozen GBM tissues to the combined effect of SDS extraction and heat-induced AR in concert with the exceptionally sensitive CIEF/nano-RPLC separations coupled with ESI-LTQ-MS/MS proteome strategy.
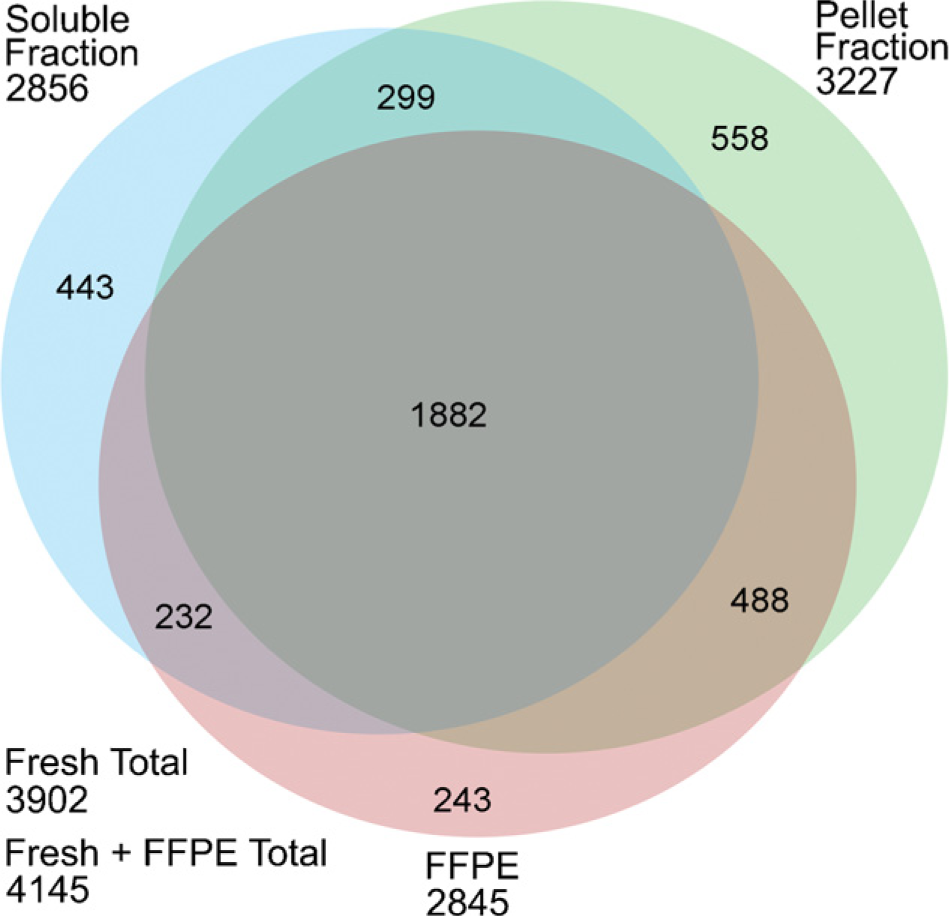
The overlap in the proteins identified from micro-dissected fresh-frozen (the soluble and pellet fractions) and FFPE GBM tissue specimens from the same patient using combined CIEF/nano-reversed-phase liquid chromatography separations coupled with nano-electrospray ionization-linear ion trap-tandem mass spectrometry.
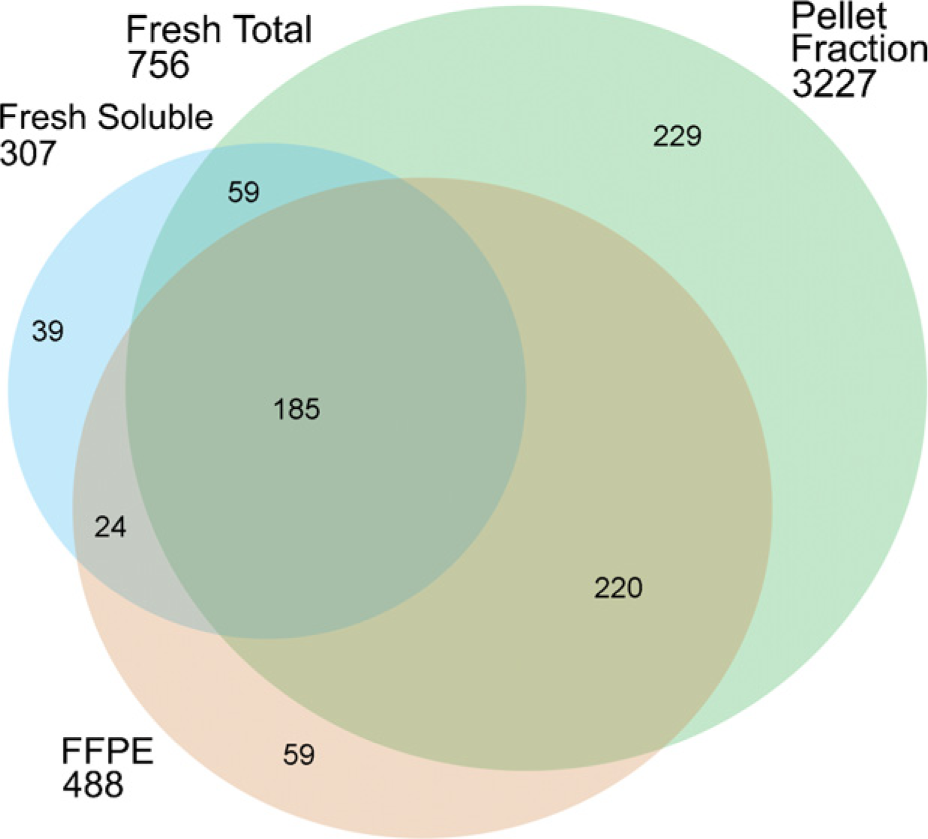
The overlap in the proteins identified from microdissected fresh-frozen (the soluble and pellet fractions) and FFPE GBM tissue specimens predicted to contain at least one or more transmembrane domains using TMHMM (www.cbs.dtu.dk/services/TMHMM-2.0/).
Discussion
Based on the heat-induced and SDS-based AR technique, minute proteins extracted from the microdissection-procured FFPE GBM tissue specimen were processed and analyzed using combined CIEF/nano-RPLC separations coupled with nano-ESI-LTQ-MS/MS. By using a decoyed-database search approach (Elias et al. 2005; Rudnick et al. 2005), an E-value threshold of 0.17, corresponding to 1% false positive of total peptide identifications, was chosen as a cutoff in this study. A total of 14,478 distinct peptides were therefore detected, leading to the identification of 2733 non-redundant SwissProt protein entries from a single proteome analysis of micro-dissected FFPE tissue.
Due to the ability of the CIEF-based multidimensional separation platform to achieve ultrahigh resolution of minute protein digests, our results present the largest catalog of proteins from a single microdissected FFPE tissue specimen reported to date. By comparison, in FFPE tissue proteome studies reported recently (Crockett et al. 2005; Hood et al. 2005; Palmer-Toy et al. 2005), the application of only a single-dimension separation because of sample amount constraints has significantly limited the capability of mining deeper into the tissue proteome.
A total of 2370 FFPE tissue proteins, or 83% of total protein identifications, overlapped with those measured from the pellet fraction of fresh-frozen GBM tissue from the same patient. This large degree of protein overlapping is the result of SDS extraction employed in both the cell pellet preparation protocol and the AR technique. The presence of SDS was critical in several AR protocols for achieving satisfactory protein extractions from FFPE tissue sections followed by IHC and SDS-PAGE analysis (Ikeda et al. 1998; Yamashita and Okada 2005). Future studies will further explore the boundaries of sample procurement, preparation, and analysis by examining how many cells must be micro-dissected to perform the analysis described in a reproducible and robust manner.
Footnotes
Acknowledgements
We thank the National Cancer Institute (CA103086 and CA107988) and the National Center for Research Resources (RR021239 and RR021862) for supporting portions of this research. This research was also supported in part by the Melvin Burkhardt chair in neurosurgical oncology and the Karen Colina Wilson research endowment fund within the Brain Tumor Institute at the Cleveland Clinic Foundation.