
Abstract
Despite the common use of immunohistochemistry in autopsy tissues, the stability of most proteins over extended time periods is unknown. The robustness of signal for 16 proteins (MMP1, MMP2, MMP3, MMP9, TIMP1, TIMP2, TIMP3, AGER, MSR, SCARB1, OLR1, CD36, LTF, LGALS3, LYZ, and DDOST) and two measures of advanced glycation end products (AGE, CML) was evaluated. Two formalin-fixed, paraffin-embedded human tissue arrays containing 16 tissues each were created to evaluate 48 hr of autolysis in a warm or cold environment. For these classes of proteins, matrix metalloproteinases and their inhibitors, scavenger receptors, and advanced glycation end product receptors, we saw no systematic diminution of signal intensity during a period of 24 hr. Analysis was performed by two independent observers and confirmed for a subset of proteins by digital analysis and Western blotting. We conclude that these classes of proteins degrade slowly and faithfully maintain their immunohistochemistry characteristics over at least a 24-hr time interval in devitalized tissues. This study supports the use of autopsy tissues with short postmortem intervals for immunohistochemical studies for diseases such as diabetic vascular disease, cancer, Alzheimer's disease, atherosclerosis, and other pathological states. This manuscript contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
Protein degradation during tissue autolysis is variable and is known to unequally affect different protein classes. Proteins have been shown to undergo specific and nonspecific proteolysis, dephosphorylation, and other changes during a prolonged postmortem interval (Fountoulakis et al. 2001). At one extreme, phosphory-lated proteins rapidly dephosphorylate in devitalized tissues, negating the utility of phosphorylation-specific antibody markers (Li et al. 2003; Warby et al. 2005). At the other extreme, cell adhesion molecules were shown to maintain comparable staining intensity during 3 days of autolysis in explanted cardiac tissues kept at room temperature (Noutsias et al. 1999). A number of lymphoid markers were evaluated in frozen and formalin-fixed tissues autolyzed out to 72 hr, before the high-temperature antigen-retrieval (HTAR) era, and showed wide variability beyond 12 hr (Pelstring et al. 1991). Most other classes of proteins have not been evaluated.
Matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) are classes of proteins involved in the regulation and turnover of extracellular matrix (Visse and Nagase 2003; Malemud 2006). Under physiological conditions they are important regulators of development, tissue repair, and tissue homeostasis. In disease states they have been targeted as potential mediators of malignancies, arthritis, fibrosis, and vascular disease. MMPs have been studied in autopsy tissues by IHC, but it was not determined to what extent MMPs degrade over a postmortem interval (Akiyama et al. 2006).
Scavenger receptors are generally regarded as proteins that bind and degrade a variety of ligands including low-density lipoproteins (LDL), oxidized LDL (oxLDL), advanced glycation end products (AGEs), β-amyloid fibrils, and bacteria (Murphy et al. 2005). Several scavenger receptors including lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1, OLR1), scavenger receptor A (SR-A, MSR), and thrombospondin receptor (CD36) are altered in atherosclerosis (Horiuchi et al. 2003; Mehta et al. 2006). IHC has been used to study scavenger receptors in autopsy tissues but without reference to staining robustness over the postmortem time interval (Kume et al. 1995).
Advanced glycation end product receptors bind AGEs, ligands known to be elevated in diabetes and renal failure (Kim et al. 2005). Receptor for advanced glycation end products (RAGE, AGER), the prototypical AGE receptor, has also been studied in autopsy tissues by IHC, without any reported differences in staining intensity as compared to surgical specimens (Cheng et al. 2005).
Despite the use of many of these proteins in IHC studies on autopsy tissues, no systematic evaluation has been performed to determine if postmortem interval influenced staining intensity, which could lead to unreliable data. We hypothesized that these aforementioned classes of proteins would maintain robust IHC signaling during at least 24 hr, if not 48 hr, of autolysis in vascular tissues.
As a surrogate to postmortem interval, we created two arrays of human surgical tissues, which were allowed to autolyze out to 48 hr. Staining patterns were then evaluated for each protein of interest to determine if there was a time-dependent loss of staining intensity. Intensities were compared by both standard pathologist scoring and by digital analysis of staining for a subset of proteins. Western blots were performed from duplicate tissue samples to correlate with the IHC staining patterns. The nonspecific TUNEL assay was used to determine if these tissues autolyzed comparably to time-matched autopsy tissues (Grasl-Kraupp et al. 1995; Tateyama et al. 1998).
Materials and Methods
Tissue Collection
Tissues were collected anonymously from eight surgical specimens, and the use of surgical pathology specimens was approved by the Johns Hopkins University Internal Review Board. Two specimens each of placenta, kidney, coronary artery, and dorsalis pedis artery were taken. Upon receiving an organ from an operating room, a block of tissue unrelated to the diagnostic material was taken. This represented time point 0, and a piece of this tissue block was sectioned off and immediately placed in formalin. The remainder of the tissue block was placed in a small closed biohazard bag and either left at room temperature (20C) or placed in a refrigerator at 4C. Portions of the tissue block were repeatedly cut off and placed in formalin at time points 12, 24, and 48 hr. All tissues, ~3–6 mm3 in dimension, were fixed informalin for a minimum of 24 hr. Collected tissues were arrayed by embedding multiple tissues in the same paraffin block to make two paraffin-embedded 16-piece arrays, one “warm” (20C) array and one “cold” (4C) array of collected tissues. A representative IHC-stained slide cut from the “warm” array is shown in Figure 1.
For Western blotting, kidney and placental tissue collections were repeated as described above, but with the pieces of tissue being snap frozen in liquid N2 and stored at −80C, rather than being formalin fixed.
IHC
IHC was performed using standard protocols. Briefly, slides were cut from formalin-fixed, paraffin-embedded tissues. After deparaffinization and rehydration, antigen retrieval was performed with most antibodies (Table 1) in a 0.01 M sodium citrate buffer (pH 6.0) in a microwave oven for 10 min. Slides were then washed (PBS) and treated with 3% H2O2 for 10 min and washed again. Slides were serum blocked and then incubated with primary antibody of differing lengths and temperatures (Table 1). Slides were washed and incubated with biotinylated anti-rabbit, anti-mouse, or anti-goat secondary antibody, depending on the primary antibody, for 30 min at room temperature (R&D Systems; Minneapolis, MN). Slides were then washed and incubated with high-sensitivity streptavidin-horseradish peroxidase conjugate (HSS-HRP) for 30 min. Slides were washed and incubated with the DAB chromogen method for visualization (R&D Systems). Slides were then counterstained with hematoxylin and coverslipped. A list of antibodies, the commercial vendor, use of antigen retrieval, and the dilutions used are listed in Table 1. All antibodies were tried at multiple dilutions on control surgical pathology tissues known to stain positive for the given antibody. Once a proper protocol and dilution were chosen, IHC was performed on the “warm” and “cold” array.
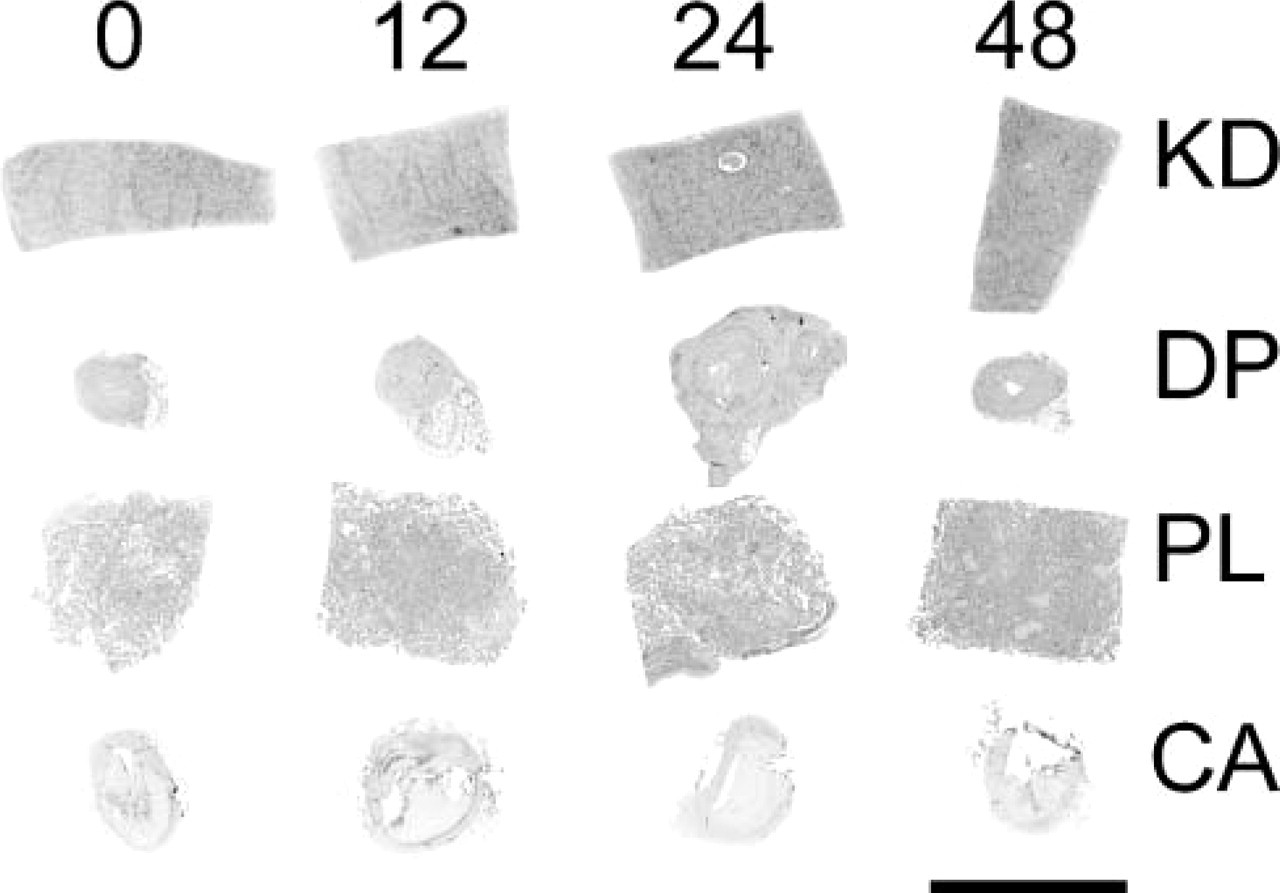
Image of a tissue array slide. Sixteen tissues are arrayed. Time point 0 hr for all four tissues is seen on the left, followed by time points 12, 24, and 48 hr, respectively. Pieces of kidney (KD), dorsalis pedis artery (DP), placenta (PL), and coronary artery (CA) are noted from top to bottom. Bar = 5 mm.
List of antibodies used for immunohistochemistry
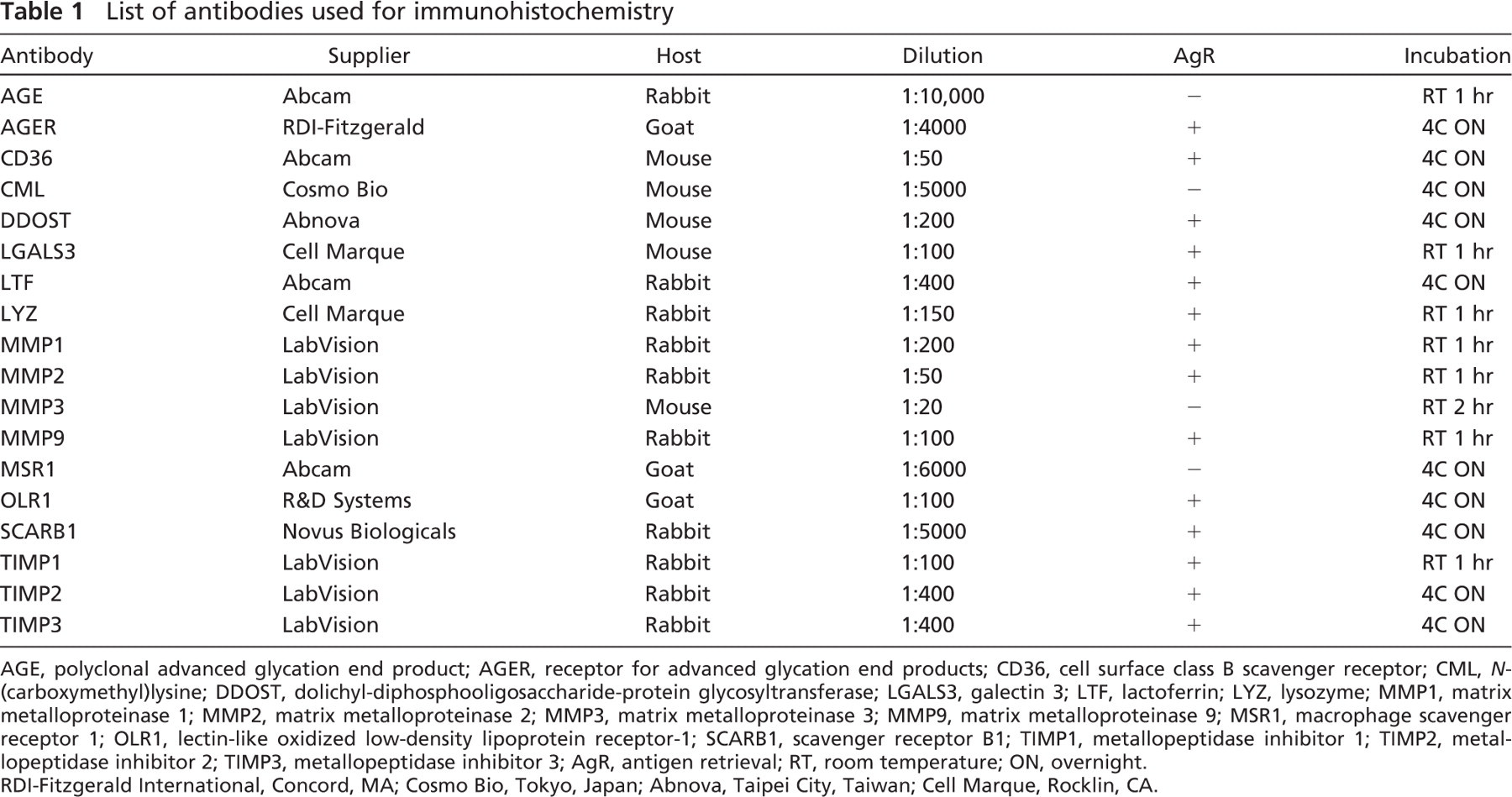
AGE, polyclonal advanced glycation end product; AGER, receptor for advanced glycation end products; CD36, cell surface class B scavenger receptor; CML, N-(carboxymethyl)lysine; DDOST, dolichyl-diphosphooligosaccharide-protein glycosyltransferase; LGALS3, galectin 3; LTF, lactoferrin; LYZ, lysozyme; MMP1, matrix metalloproteinase 1; MMP2, matrix metalloproteinase 2; MMP3, matrix metalloproteinase 3; MMP9, matrix metalloproteinase 9; MSR1, macrophage scavenger receptor 1; OLR1, lectin-like oxidized low-density lipoprotein receptor-1; SCARB1, scavenger receptor B1; TIMP1, metallopeptidase inhibitor 1; TIMP2, metal-lopeptidase inhibitor 2; TIMP3, metallopeptidase inhibitor 3; AgR, antigen retrieval; RT, room temperature; ON, overnight. RDI-Fitzgerald International, Concord, MA; Cosmo Bio, Tokyo, Japan; Abnova, Taipei City, Taiwan; Cell Marque, Rocklin, CA.
Protein Purification
Frozen human tissue samples were thawed and homogenized on ice in lysis buffer (Tris-HCl, pH 7.5, 50 mM, NaCl 150 mM, SDS 0.1%, deoxycholate 0.5%, NP-40 1%) with protease inhibitors using a Brinkman Polytron homogenizer (Brinkmann Polytron; Westbury, NY). The homogenate was centrifuged at 4C to separate cell debris, and protein extract concentrations were determined by BCA protein assay reagent (Pierce Biotechnology; Rockford, IL). All reagents were purchased from Sigma Chemical Co. (St Louis, MO) unless otherwise stated.
Western Blotting
Western blotting was performed on proteins purified from each of the time points for both kidney and placental tissues. Total protein (60 μg/lane) was boiled with 2X SDS-PAGE loading buffer (100 mM Tris, pH 6.8, 4% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.2% bromophenol blue) and separated in a 4–12% NuPAGE Novex Bis-Tris Gel (Invitrogen; Carlsbad, CA) for each sample and time point. The resolved proteins were electrotransferred onto 0.45-μm PVDF membranes. After blocking in 4% milk-PBST solution, the membrane was sequentially incubated with primary antibody and HRP-conjugated secondary antibody. The antibodies tested include anti-MMP2 (0.4 μg/ml; Lab Vision, Fremont, CA), anti-SCARB1 (1 μg/ml; Novus Biologicals, Littleton, CO), anti-TIMP1 (0.4 μg/ml; Lab Vision), and anti-TIMP2 (0.4 μg/ml; Lab Vision). Antibody to β-actin (0.1 μg/ml; Abcam, Cambridge, MA) was used as a control for equal loading. Immunoreactivity was detected using the SuperSignal West Femto Maximum Sensitivity Substrate (Pierce). Protein levels were evaluated for robustness of signal and band-size alterations across each of four time points (0, 12, 24, 48 hr). Optical densitometry and area under the curve (AUC) analysis was performed using ImageJ for each protein band (Wayne Rasband, National Institutes of Health, <http://rsb.info.nih.gov/ij/>; ImageJ is in the public domain).
Measurement of Apoptosis
TUNEL assay was used to determine the degree of autolysis. Briefly, formalin fixed, paraffin-embedded slides were deparaffinized, washed, incubated with proteinase K (5 mg/ml; Dako, Fort Collins, CO) for 15 min, washed, and then bathed in H2O2 for 5 min. Slides were then incubated with terminal deoxynucleotidyl transferase (TdT) for 1 hr at 37C, washed, and then incubated with anti-digoxigenin peroxidase conjugate reagent for 30 min according to the manufacturer's instructions (ApopTag Plus Peroxidase In Situ Apoptosis Kit; Millipore, Billerica, MA). After washing, slides were stained with a DAB peroxidase substrate kit (Vector Laboratories; Burlingame, CA), counterstained with eosin Y (Richard-Allan Scientific; Kalamazoo, MI), and coverslipped.
Pathologists' Review
Each slide was reviewed independently by two pathologists (JM and MKH). Each tissue was graded on a 0–3 scale, with 0 being no staining and 3 being intense staining. When tissue was missing on a slide or was not able to be evaluated, it was removed from analysis. This initial score was termed the absolute staining intensity (ASI) value and was measured as 0, 1, 2, or 3. It was determined that, in some tissues, staining was strong (2–3), but in other tissues staining was weak (0–1). As the measure of greatest interest was a change in staining intensity over the time course, these ASI measures (0, 1, 2, 3) were converted into relative staining intensity (RSI) values (-2, −1, 0, 1, 2) for each sample. This novel grading method allowed cross analysis between samples with different starting ASI to be normalized. To make this conversion, for any given antibody and tissue the most common ASI across all four time points was given the RSI value of 0. Then all other values for that antibody and tissue were relatively scored. For example, ASI scores of 3, 2, 2, and 1 at time points 0, 12, 24, and 48 hr, respectively, in the kidney sample of LYZ were converted to RSI scores of 1, 0, 0, and −1 (Figure 2). If two tissues stained at a high ASI and two tissues stained at a lower ASI (e.g., 3, 3, 2, 2), the conversion of ASI into RSI was made to show a loss of signal (e.g., 0, 0, −1, −1). To determine percent deviation of RSI for each antibody at each time point, the sum of deviated RSI values was divided by the sum of all tissues evaluated at that time interval. Each evaluated slide contained up to 16 tissues, but some tissues were lost during processing on certain slides.
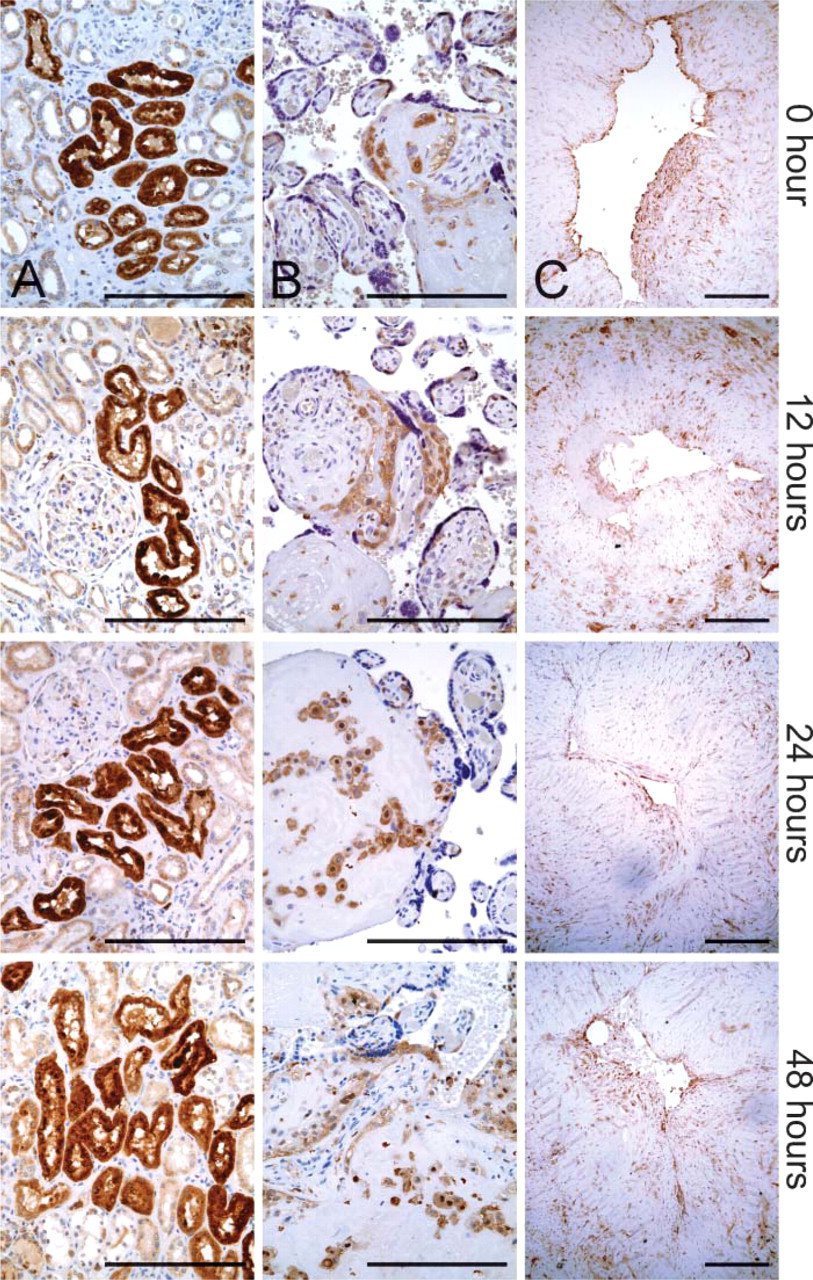
Representative images of immunohistochemistry for LYZ, LGALS3, and MMP1 proteins. (
A loss of staining intensity value for each antibody over the 48-hr time course was generated. The relative loss of intensity values over all four tissues from both arrays was summed (up to 8). This value was then divided by the sum of tissues studied (up to 8). This generated values that correspond to the percent of tissues losing staining intensity, but does not indicate how great the loss of intensity was (Figure 3).
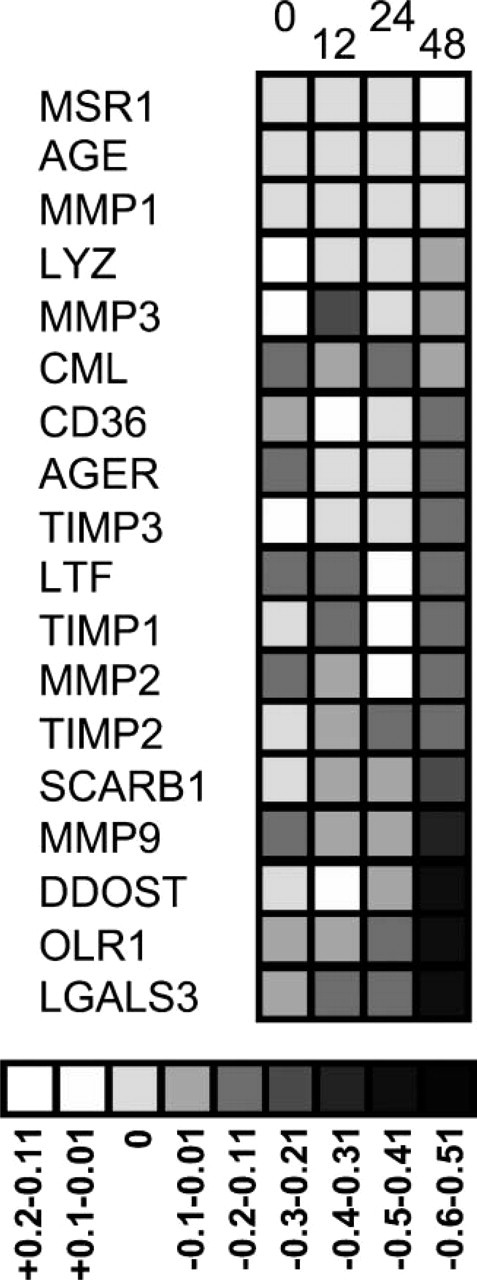
Grayscale comparison of RSI during a period of 48 hr. Staining across the four tissues has been merged for each antibody. The numbers 0, 12, 24, and 48 represent the four time points. The scale of average intensity change is at the base of the figure. Darker colors represent greater loss of staining intensity (up to a 60% loss in relative staining).
Digital Analysis of Staining
“Warm” and “cold” array slides for MMP1, MMP2, MMP3, and MMP9 were scanned on an Automated Cellular Imaging System (ACIS II) digital image analyzer (Chromavision; Aliso Viejo, CA). An arbitrary color template for antibody staining (brown) and counterstain with Meyer's hematoxylin (blue) was created, taking into account hue, luminosity, and intensity. Each tissue was individually scored for amount of brown and blue as a percent of tissue space. Brown intensity was then calculated as a percent of tissue stained (0–100% scale). Average staining intensity was calculated for each tissue over four time points, and loss of intensity was interpreted as an intensity value 10% lower than the four-tissue average.
Data Handling
All data were recorded and processed in an Excel database (Microsoft; Redmond, WA). ANOVA was performed to correlate digital and observer scoring. χ2 test was performed onthe distribution of intensity loss using the Web Chi Square Calculator (http://www.georgetown.edu/faculty/ballc/webtools/web_chi.html, Georgetown University, accessed 10/3/2006).
Results
Observer Analysis of IHC
IHC staining intensity was evaluated for 543 pieces of tissue by two observers. Both increases and decreases in staining intensity were noted, with 10% of all deviations representing a relative loss of signal intensity and 2% representing an increase in relative signal intensity. Only 3/543 tissues (<1%) had a large relative loss of staining intensity (-2), indicating that when staining loss was observed, it was predominantly a moderate loss (-1) rather than a marked loss from a previously robust staining pattern. With the exception of a loss of staining of MMP3 in the four tissues at time point 0 in the “cold” array due to a lack-of-staining artifact, there was no categorical loss of staining across all four tissues for any antibodies. These four array points were removed from all further analysis.
Relative signal intensity change during a period of 48 hr was determined for each antibody as described in Materials and Methods (Figure 3). The greatest percentage of tissues with a decrease in signal was noted at 48 hr for the proteins MMP9, DDOST, OLR1, and LGALS3 (Figure 2 and Figure 3). No proteins studied showed a relative staining loss in >20% of tissues at 24 hr. Of 18 proteins, 14 (78%) had <8% loss of intensity in stained tissues (Figure 2).
Overall loss of staining within the entire cohort of proteins was also determined. The relative staining values for each of the 18 antibodies was averaged over all four tissues in both “warm” and “cold” arrays. Analysis of all data points showed that the loss in signal intensity was greatest at 48 hr (18% of samples) and was comparable for time points 0, 12, and 24 hr (8%, 7%, 5%, respectively) (Figure 4). χ2 analysis of this distribution was significant for the loss of intensity at 48 hr only (p<0.001, χ2 = 34.01, df = 3). Intensity did vary by tissue type with more overall variation in staining intensity in the dorsalis pedis artery and the coronary artery (16% and 14%), compared with the kidney and placenta (8% and 9%). A kappa statistic of interobserver variation was 0.24, which represents fair agreement (Viera and Garrett 2005).
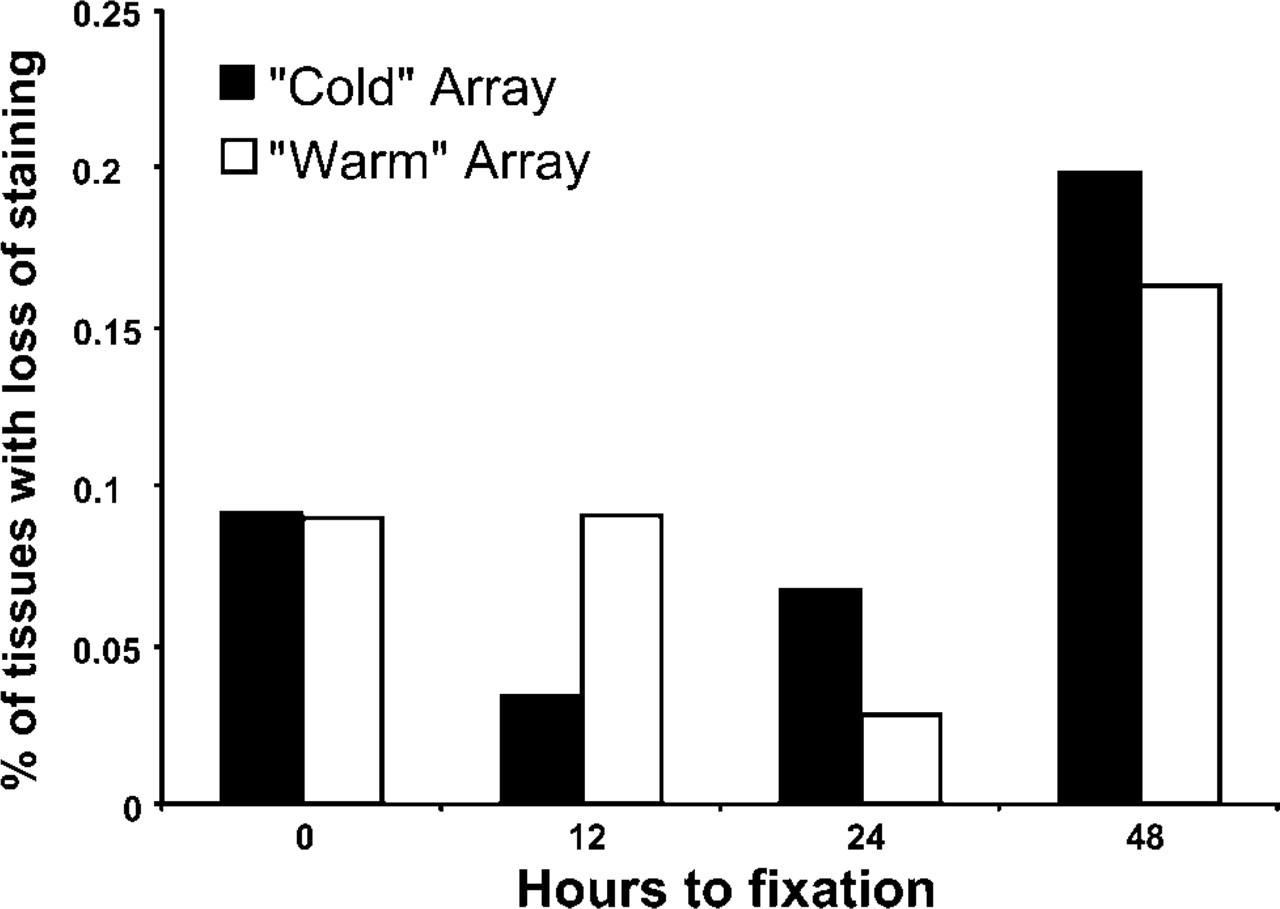
Loss of signal intensity over 48 hr of autolysis in both “cold” and “warm” arrays for all 18 antibodies. Loss is greatest at the 48-hr time point.
The four MMP IHC stains (MMP1, MMP2, MMP3, and MMP9) were evaluated as a subset of the tissues for comparison to a digital scoring method. All independent observations were combined for each time point. For the MMPs, the percent loss in signal intensity was 9%, 10%, 2%, and 14% at time points 0, 12, 24, and 48 hr across all four tissues and covering both arrays.
Digital Analysis of Staining
Analysis of a subset of slides (MMP1, MMP2, MMP3, and MMP9) was performed on a Chromavision ACIS II system. Percent brown staining values were generated for each spot that ranged from 0 to 97% intensity (mean 66%). No antibodies were shown to have time-sensitive loss of staining over all four tissues. Individual data points for each antibody were then pooled into four time points to compare signal intensity values to pathological reviewer scoring. The percent loss in signal intensity was 14%, 16%, 3%, and 13% at time points 0, 12, 24, and 48 hr across all four tissues. Correlation between digital and observer staining was very good (r 2 = 0.65) for the evaluated proteins (Figure 5).
Western Blot Analysis
Western blots were performed for four study proteins and one control protein (β-actin, MMP2, SCARB1, TIMP1, and TIMP2). Selection of evaluated proteins was based on non-overlapping protein band sizes and the reported ability of a given antibody to work for Western blotting, in addition to its primary use for IHC. Overall, protein levels remained surprisingly robust for all proteins studied in both placental and renal tissues (Figure 6). When all band intensity values generated through AUC analysis were merged for each protein across both tissues, the average loss of band intensity was 1%, 9%, and 10% at time points 12, 24, and 48 hr, respectively. At 48 hr, the signal loss ranged from 0 (TIMP2) to 25% (TIMP1). There was no obvious band-size alteration or increase in alternative bands suggesting methodical proteolysis in the devitalized tissues throughout 48 hr.
Measurement of Apoptosis
TUNEL assays identified increasing autolysis over the 48-hr time course. TUNEL was performed on both “warm” and “cold” arrays and selected autopsy tissues. At time 0, only faint positivity was observed in some renal tubules, whereas all other tissues were negative. By 24 hr, strong staining was observed in renal tubules and placenta. Renal tubular staining was comparable to that observed in the time-matched kidney autopsy tissue (Figure 7). Endothelial cells were positive for TUNEL staining by 24 hr. Vascular smooth muscle cells did not show positivity throughout 48 hr in the “warm” and “cold” arrays but were faintly positive in autopsy tissues at 23 hr. Inflammatory cells, predominantly macrophages within an atherosclerotic plaque in the coronary artery, were strongly positive at 48 hr. TUNEL staining results were similar between “warm” and “cold” arrays and tissues taken from autopsies with comparable postmortem intervals (Figure 7).
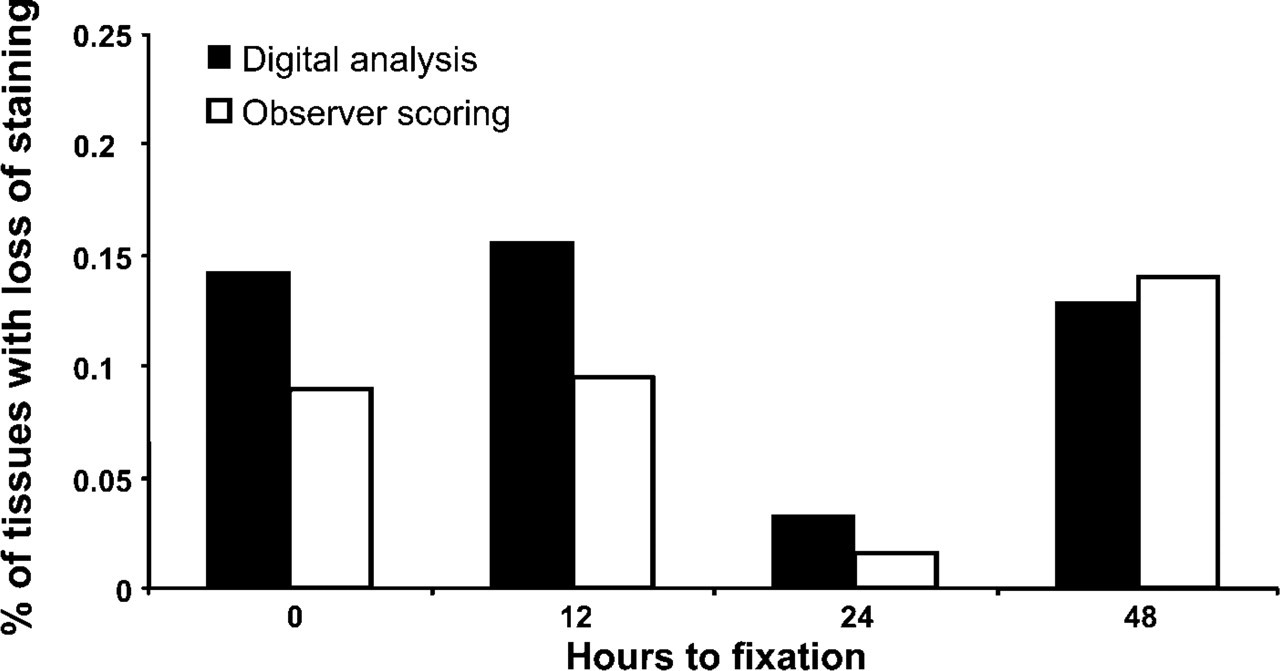
Overall digital analysis (Chromavision) vs observer scoring for MMP1, MMP2, MMP3, and MMP9. Up to 32 tissues were analyzed for each time point. There was no significant difference between the two distributions (ANOVA, p>0.05).
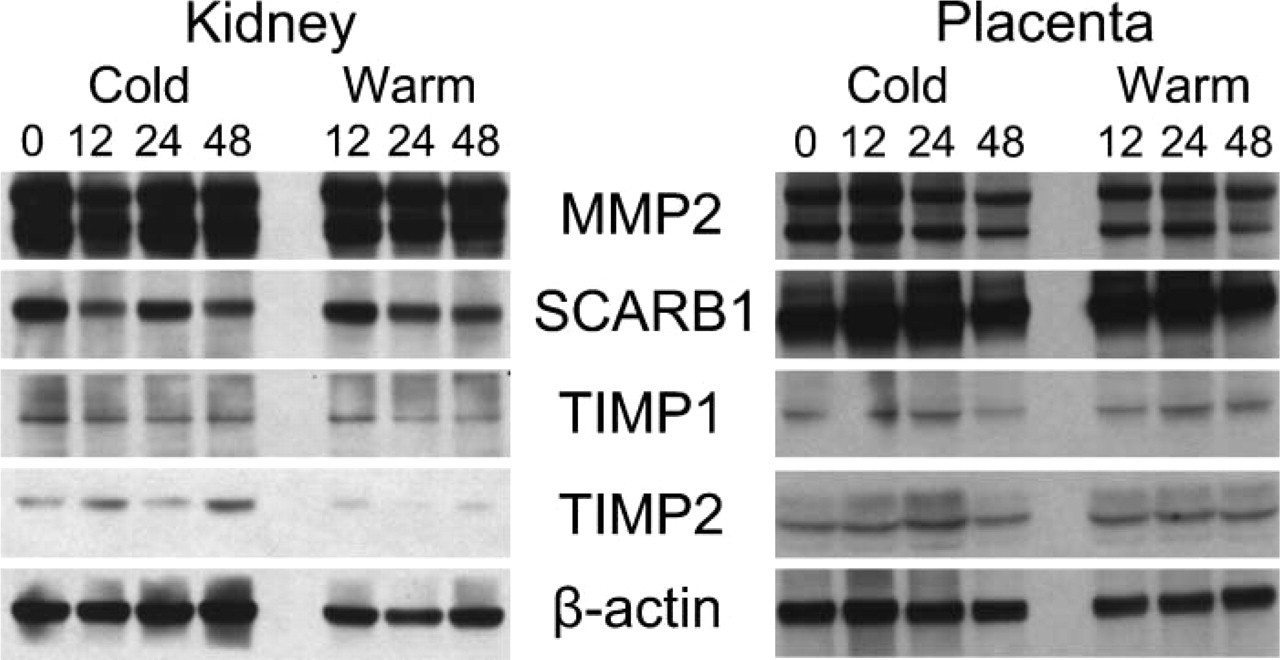
Western blots of selected proteins. Sixty μg/lane of protein was loaded in each lane. Numbers 0, 12, 24, and 48 represent time points at which tissue was frozen. Protein concentrations differed significantly for MMP2 and SCARB1 between kidney and placenta. An apparent increase in the control β-actin at 48 hr in the cold kidney appears to indicate a higher level of loading of this lane.
Discussion
This is the first systematic evaluation of IHC in autolyzed tissues for classes of proteins including MMPs and their inhibitors, scavenger receptors, AGEs, and AGE receptors. These results demonstrate that for at least 24 hr of autolysis, an apparently large number of diverse proteins remain intact and can still be quantitatively examined by both IHC and Western blot analysis. Even at 48 hr of autolysis, the decrease in most protein levels was only moderate. Variation in staining intensity was most affected by the time interval prior to tissue fixation followed by heterogeneity between cell types present and experimental artifact. Of the 18 antibodies studied, none showed a loss of staining intensity in all four tissues at 48 hr. Only when the data were summed across all antibodies was there a noticeable loss of signal intensity at 48 hr. We saw no difference between staining intensity loss between the “warm” array and the “cold” array. We interpret this finding to indicate that even tissues kept at room temperature for up to 24 hr do not undergo significant degradation of these proteins. Our TUNEL assay results confirmed the presence of autolysis over this time course in both arrays, indicating that our surgical tissues did degrade over this time course, similar to postmortem interval-matched autopsy tissues.
Although these results are encouraging regarding the use of autolyzed tissues, namely, autopsy tissues, for IHC study, they are not likely to be true for all proteins or all autopsy tissues. None of these proteins is known to be phosphorylated or to have other modifications subject to rapid reversal. They are also not within signaling pathways that rapidly turn over. Also, blood vessels represent a more stable tissue source than predominantly epithelial tissues such as the liver or pancreas, which were avoided. Observations of multiple autopsies have shown that epithelial organs autolyze more rapidly than other organs and that the vasculature (with the exception of endothelial cell sloughing) remains remarkably intact histologically over long postmortem intervals.
For each organ evaluated, all material came from a single surgical specimen and was allowed to remain in an unhydrated state, such that autolysis was unimpeded between time points. An advantage of this method, rather than a method of using tissues collected from separate autopsies at postmortem intervals of 12, 24, and 48 hr, is the expectation that staining should be equal across all four time points, and interindividual differences would not be a significant cause of staining heterogeneity. In this study, the most consistent staining was observed in the most homogeneous tissues, the kidney and placental tissues (only 8–9% variation). In the vascular tissues in which atheromatous plaques were present, the presence and amount of certain inflammatory cells differed between sections, resulting in more observed heterogeneity (14–16%). This was particularly true for some stains that predominantly mark macrophages, such as CD36 and SCARB1.
This experimental protocol was designed as a way to evaluate antibodies that can be used in autopsy materials with reasonable postmortem intervals. Although these data show that, for at least 24 hr, IHC results remain robust in autolyzed surgical tissues, it is not an exact recapitulation of a true postmortem interval.
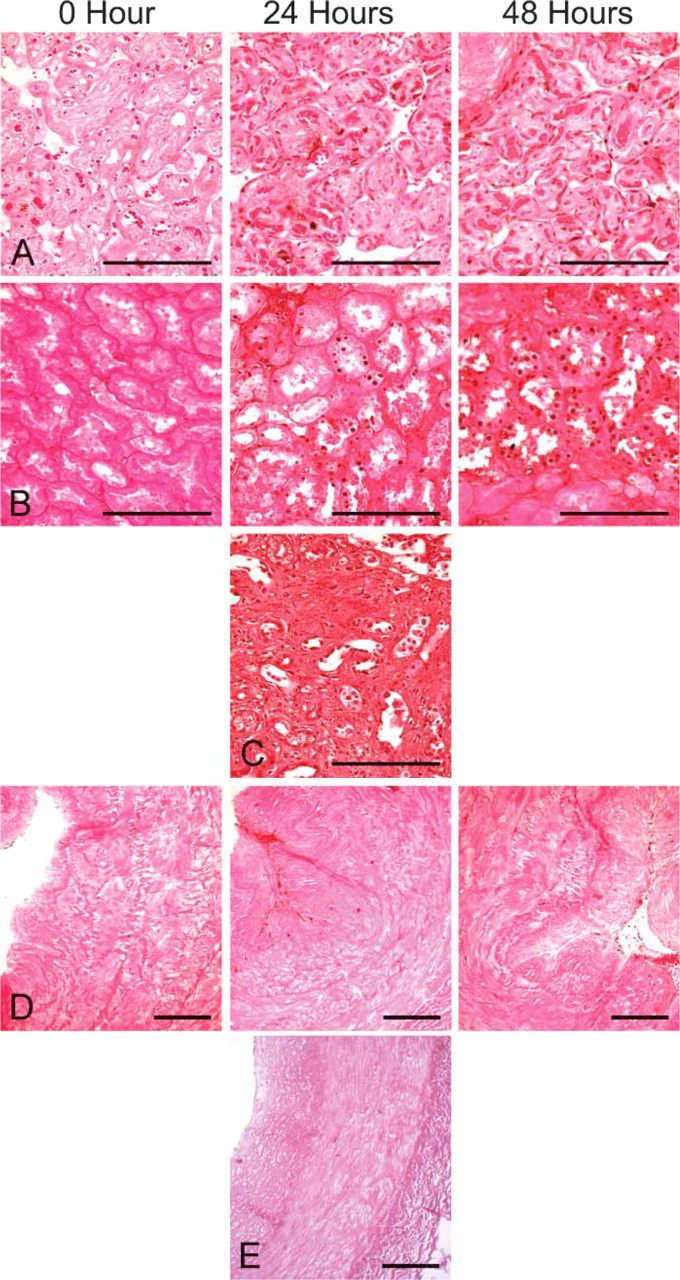
TUNEL assay results. (
The time course of cooling difference between surgical specimens and autopsy tissues represents the greatest limitation of this study. Surgical specimens rapidly cool from a body temperature of 37C to room temperature, due to their small sizes. This occurred for all of our specimens, after which we chose to keep the harvested tissues at either 4C or room temperature to recapitulate best- and worst-case scenarios of protein degradation over the remaining time course.
The cooling rates of bodies remains an inexact science, owing to a number of variables such as body surface area, insulation, microclimate, and an initial warming of the body (Hutchins 1985; Nelson 2000). One overly simplistic calculation, the “rule of thumb” for body cooling, is that a body generally cools at 1C/hr plus 3 hr (Stipanits and Henssge 1985; Henssge 1988). Therefore, a body left at room temperature would take ~20 hr to cool from 37C to an ambient temperature of 20C. This general measurement is not as useful for hospital deaths where bodies are quickly moved to a refrigerated morgue (4C) prior to autopsy. Body cooling in this colder environment would likely drop core body temperatures more rapidly, although the exact effect of a cold-storage morgue on cooling acceleration has not been shown. Additionally, not all tissues collected at autopsy will be subjected to the same pace of cooling. For example, thyroid tissue should be cooled more rapidly, whereas more centrally located tissues in the body core, such as the liver, would cool more slowly and therefore would autolyze more significantly.
We have initiated an IHC study of these same antibodies in autopsy subjects with postmortem intervals out to 28 hr. There is increased autolysis in the autopsy tissues with longer postmortem intervals as determined by histological criteria on hematoxylin/eosin-stained tissues and by TUNEL assay. By histological criteria only, there is more autolysis in core body tissues of autopsy subjects, relative to our “warm” and “cold” arrays at matched time points. Conversely, IHC staining among autopsy subjects is robust, although variable between tissues and individuals, as expected. There is no correlation between postmortem interval and staining intensity in the evaluated antibodies in our collection of autopsy tissues. Examples of staining comparable to that seen on the “warm” and “cold” arrays are provided (Supplemental Figure 1).
Due to the difference in histologies including renal tubule vacuolization and endothelial sloughing between time points 0 and 48 hr, it was not possible to blind the pathologists in this study. Therefore, observer bias could have affected these results. However, the digital scoring method (ACIS), which was unbiased, showed the same degree of staining variability in the MMP slides. This agreement (r 2 = 0.65) was a useful validation of our observer scoring methods. Before removing the MMP3 “cold” array values at time 0, due to a lack of staining artifact the agreement was even stronger (r 2 = 0.87). As tissue microarray systems and automated scoring methods become more robust, pathologist observer scoring methods will likely lose favor, and digital methods will become the standard, further reducing the possibility of observer bias (Chen et al. 2004; Fedor and De Marzo 2005; Eguiluz et al. 2006).
Western blot data provided a second line of evidence of protein viability over this time period, supporting and augmenting IHC results. The intact nature of the proteins shown on Western blot indicates that not only are the specific epitopes recognized by the antibodies still present in the tissue as seen by IHC, but there is apparently little protein degradation at all. Robust Western blot data from autopsy tissues collected out to 41 hr from a study of perinatal human lungs has been reported (De Paepe et al. 2002). Consistent with that report, we found nearly equal protein band intensity in all our samples out at least 24 hr.
Our findings indicate that formalin-fixed, paraffin-embedded archival autopsy materials with short postmortem intervals represent a largely untapped source of materials to study human disease. There is a broad range of pathologies associated with the aforementioned protein classes. MMPs and their inhibitors have been widely studied in malignancies (Fingleton 2006). Generally, multiple metastases in numerous organs are not removed from a given individual during any standard surgical procedure. The role of MMPs in the meta-static spread of tumor may be best studied by using archival autopsy tissues taken from individuals who have succumbed to widely metastatic cancers (Embuscado et al. 2005).
Scavenger receptors are frequently reported to be present in inflammatory cells in atherosclerotic plaques (Murphy et al. 2005; Moore and Freeman 2006). Correlating the distribution of the myriad of scavenger receptors in different atherosclerotic plaque types in human coronary arteries can easily be studied from archival autopsy heart sections. Advanced glycation end-product receptors have more recently been strongly implicated in diabetic vascular disease and Alzheimer's disease (Stern et al. 2002). Human Alzheimer's disease research is strongly dependent on the study of whole brains collected at autopsy and represents a ready source of tissues (Mikolaenko et al. 2005). The study of AGEs and their receptors in human diabetic vascular disease has been performed in a piecemeal fashion, generally one vascular bed per study (Sakata et al. 1998; Tanji et al. 2000). It would be useful to collect a wide range of vascular beds from the same individual to understand the role of AGEs and AGE receptors in macro-and microvascular diabetic vascular disease globally. This cannot be achieved from surgical specimens.
In conclusion, we have demonstrated that tissues subjected to conditions generally considered to develop autolysis do maintain robust IHC staining intensity and protein stability for at least 24 hr for certain classes of proteins. This study challenges the traditional belief that autopsy tissues are not valuable for protein studies. Our data suggest that autopsy tissues with postmortem intervals up to 24 hr can be used for IHC studies for these classes of proteins.
Footnotes
Acknowledgements
This work was supported by the American Diabetes Association (1-05-JF-20 to MKH) and by National Institutes of Health Grant P01 HL-056091 (to KFT).
The authors thank Drs. Angelo De Marzo and William Baldwin III for helpful comments on the manuscript and the use of a digital camera. The authors also thank Kristen Lecksell for assistance with the digital analysis.