
Abstract
After immunohistochemistry (IHC) began to be used routinely, a number of investigators worked on methods for staining multiple molecules in the same tissue sections or cells. Achieving this goal was not easy, however. One reason for this is that the majority of primary antibodies used in IHC reactions are raised in rabbits, and recognizing signals from two different rabbit antibodies is not trivial. Thus, all of the protocols described to date have serious limitations. Here we report a simple, quick, and inexpensive solution to the problem. It has two major advantages over existing methods. First, by using antibodies from the same host, two or more antigens can be visualized in the same section with commercially available fluorescent dyes. Second, because the technique relies on signal amplification, both rare and abundant antigens can be detected.
Materials and Methods
Animals
Adult male mice and rats were terminally anesthetized and perfused transcardially with 4% paraformaldehyde. Ileums and brains were removed and submerged in 10% and then 20% sucrose solutions (15–18 hr each). This served to cryo-protect the tissues, which were next quickly frozen on dry ice, wrapped in aluminum foil, and stored at −80C until they were sectioned. Ten-μm-thick sections were cut in a cryostat, thaw mounted on Super Colorfrost/Plus slides (Fisher Scientific; Pittsburgh, PA), and kept at −80C until they were stained.
Microwave Treatment (MWT) and IHC
MWT for Double IHC. Slides were placed flat on the bottom of a plastic container (12 × 9 × 6 cm) filled with 200 ml of 10 mM citric acid buffer, pH 6.0. An 800-W household microwave oven was used to first bring the liquid to the boiling point (~2.5 min) at 100% power, and then the sections were microwaved for an additional 5 min at 50% power. Slides were allowed to cool in the citric acid buffer for 30 min at room temperature and then rinsed with 1X PBS.
Endogenous Peroxidase Activity. Because TSA is based on a chemical reaction catalyzed by peroxidase (HRP) enzyme, it is essential to be sure that there is no tissue-derived (endogenous) HRP activity that would result in a nonspecific reaction. To make sure that we could visualize and eliminate such activity, we used rat small intestine as a test tissue because white blood cells in the lamina propria are well known for exhibiting HRP activity that is very hard to block (Hunyady et al. 2000). Sections of perfused rat ileum were washed three times for 3 min each in PBS and then one time in Tris-HCl, pH 8.0. Endogenous peroxidase activity was visualized with a DAB reaction using reagents from the SuperPicture HRP Polymer conjugate rabbit primary (DAB) kit (cat. #87-9263; Zymed Laboratories, South San Francisco, CA). HRP activity in the tissue could be eliminated by microwaving sections for ~2.5 min in 10 mM citric acid buffer, pH 6.0, at 100% power (until the liquid starts to boil) and then letting the slides cool in the same buffer for 30 min at room temperature.
IHC. Coronal sections at the levels of the hypothalamus and striatum of mouse and rat brains were stained. Slides were washed three times (for 3 min each) in PBS and subsequently incubated for 10 min in a 1X Universal Blocking Reagent (Biogenex; San Ramon, CA). They were then rinsed with distilled water and incubated in the primary antibody. All antibodies were diluted to working concentrations in PBS containing 1% BSA and 0.25% Triton X-100. When the TSA reaction was performed later, endogenous peroxidase activity was blocked by traditional 15-min incubation in a 3% hydrogen peroxide solution for comparison purposes with sections that underwent MWT. For detection we used a variety of reagents described below. Between incubations we washed the sections three times in PBS (3 min per wash). The following staining methods were used.
Testing the Effect of MWT on Fluorescence Intensity of a Previous Staining (Vasopressin Neurophysin IHC). The primary antibody, PS 41, diluted to 1:50 (mouse monoclonal provided by H. Gainer; NIH, Bethesda, MD) was applied for 1 hr at room temperature and developed using three different techniques. In the first two techniques, an affinity-purified anti-mouse IgG antibody was used that was conjugated to either an AlexaFluor 488 (1:1000, donkey; Invitrogen, Carlsbad, CA) or to a biotin-SP (1:1000, donkey; Jackson ImmunoResearch Laboratories, West Grove, PA). The latter was then visualized with AlexaFluor 488 conjugated to streptavidin (1:1000; Invitrogen). All incubations were performed for 1 hr at room temperature. A third technique utilized the SuperPicture HRP Polymer conjugate for mouse primary antibodies (Zymed Laboratories). The conjugate was applied to sections undiluted for 30 min at room temperature, followed by a Tyramide-FITC (cat. #SAT701B001EA, TSA Fluorescein Tyramide Reagent Pack; PerkinElmer Life and Analytical Sciences, Boston, MA) reaction according to the manufacturer's instructions.
Testing the Ability of MWT to Eliminate Cross-reactivity Between IgGs Raised in the Same Host. For these tests, double IHC was used with a microglia-specific antibody (Iba) in combination with either tyrosine hydroxylase (TH) or orexin (Figure 1). Duplicate slides were prepared for primary incubations to compare treatments. Sections were incubated first in anti-Iba (1:1000, rabbit; Chemicon, Temecula, CA) overnight at 4C, then in biotin-SP-conjugated anti-rabbit IgG raised either in donkey (1:1000; Jackson ImmunoResearch Laboratories) for the double staining with TH (1:500, rabbit; Chemicon) or in goat (1:1000; Vector Laboratories, Burlingame, CA) for double staining with orexin (1:50, goat; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 hr at room temperature. Next, slides were incubated in AlexaFluor 488-conjugated streptavidin (1:1000; Invitrogen) for 1 hr at room temperature. One group of slides was microwave treated before applying the anti-TH or the anti-orexin primary antibodies overnight at 4C. The second stainings were visualized by AlexaFluor 594-conjugated anti-rabbit IgG (1:1000, goat; Invitrogen) and anti-goat IgG (1:500, donkey; Invitrogen) secondary antibodies for 1 hr at room temperature, respectively.
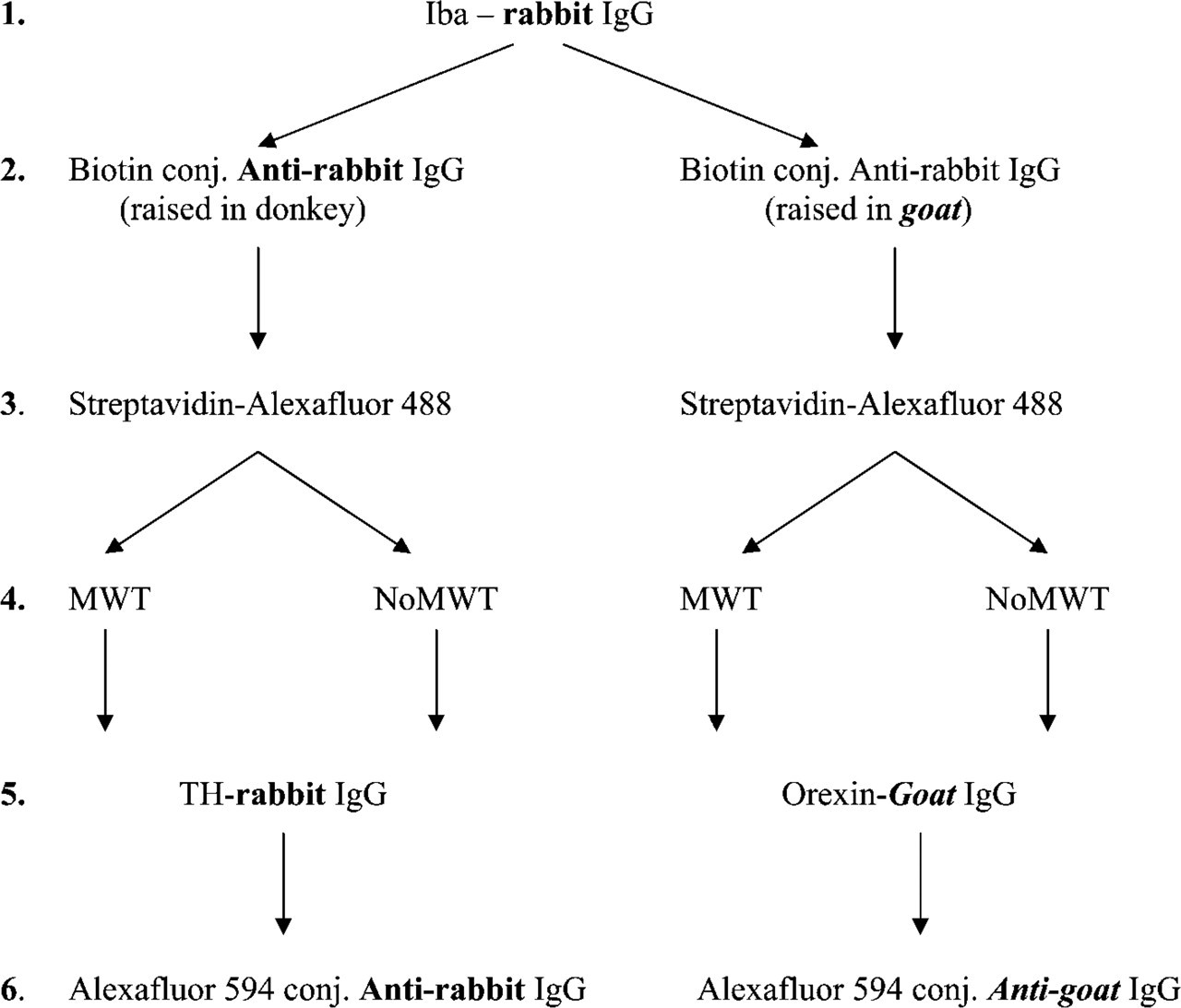
Step by step illustration of the method applied to test the effect of microwave treatment (MWT) on fluorescent double immunostainings performed using antibodies raised in the same species. The potential cross-reactivity problems that might result in false colocalization signals are indicated by bold characters. Iba: microglial marker, TH: tyrosine hydroxilase.
Testing the Effect of MWT When Amplifying Both Signals and Using Antibodies From the Same Host. Double IHC was performed using a combination of a neuronal-specific nuclear antibody, NeuN, and a TH antibody (Figure 2). The anti-NeuN (1:200, mouse; Chemicon) was applied for 1 hr at room temperature, followed by incubation in SuperPicture HRP Polymer conjugate for mouse primary antibodies (Zymed Laboratories) and AlexaFluor 594-Tyramide (Invitrogen). After the first immunostaining, one group of slides was microwave treated. Slides were then incubated in anti-TH primary antibody (1:1000, mouse; Incstar Corporation, Stillwater, MN) for 1 hr at room temperature. The second immunostaining was developed again using the SuperPicture HRP Polymer conjugate for mouse primary antibodies, and FITC-Tyramide (PerkinElmer) was used as the fluorochrome for visualization.
A combination of TH, the rate-limiting enzyme in catecholamine biosynthesis (an accepted marker of dopaminergic neurons) and GFAP (an astrocyte-specific marker) was used to perform double IHC (Figure 3). For the first immunostaining, an anti-TH antibody was used here that was raised in rabbit (1:500; Chemicon) and an HRP-conjugated anti-rabbit IgG (1:100, goat; Vector Laboratories) was used as a secondary antibody (1 hr and 2 hr incubations at room temperature, respectively). Staining was then developed by FITC-Tyramide according to the manufacturer's instructions. After MWT, an anti-GFAP antibody (1:500; Dako, Carpinteria, CA) that was also raised in rabbit was applied for 1 hr at room temperature. Secondary detection was the same as for the TH immunostaining, except that AlexaFluor 594-Tyramide (Invitrogen) was used for the visualization.
To demonstrate the power of the technique, we performed a triple immunostaining using same host (rabbit) antibodies. Vasopressin, CRF, and TH immunostaining were performed on the same section from colchicine-treated mouse (Figure 4). Vasopressin antibody (VA4, rabbit) (Alstein et al. 1988) was used at a 1:1000 dilution for 2 hr, the anti-TH antibody (rabbit, cat. #AB 151; Chemicon) was used at 1:500 for 1 hr, and the anti-CRF (a gift from Wylie Vale) (Sawchenko and Swanson 1981) was used at 1:5000 for 3 hr, all at room temperature. Endogenous peroxidase blocking was performed after the incubation with the first primary (vasopressin) antibody. Immunostainings were developed using SuperPicture HRP Polymer conjugate for rabbit primary antibodies (Zymed Laboratories), FITC-Tyramide (PerkinElmer) for the vasopres-sin, AlexaFluor 568-Tyramide for the CRF, and CY5-Tyramide for the TH (all from PerkinElmer). We applied a MWT step after each immunostaining.
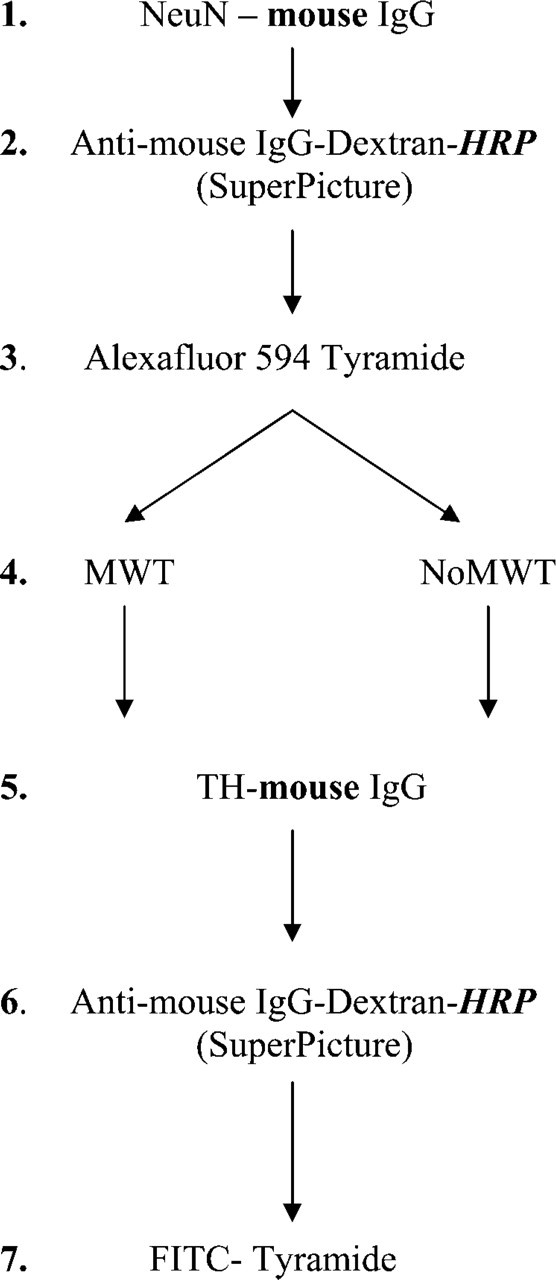
Step by step illustration of the method applied to test the effect of MWT on NeuN (neuronal cell marker) and TH fluorescent double immunostaining performed using primary antibodies from the same host animals (mouse) and amplification of the signals. We used the same anti-mouse HRP Polymer conjugate (SuperPicture) as secondary antibody for both stainings. The potential species and peroxidase enzyme (HRP) cross-reactivity problems that might result in false colocalization signals are indicated by bold characters.
All immunostainings were evaluated using a conventional inverted Leica microscope (DMI 6000B; Leica, Wetzlar, Germany) equipped with the appropriate fluorescent filter cubes (A4, TX2, and L5) using an EXFO illumination system (EXFO; Mississauga, Ontario). The microscope is driven by an Apple computer (G4; Apple, Cupertino, CA) using Volocity software (Improvision Inc.; Lexington, MA) for image capturing.
Results
MWT as described above completely eliminates endogenous peroxidase activity in the rat ileum (Figure 5). Without MWT, many cells (shown by arrows) are stained by DAB (Figure 5A). After the section has been microwaved, there is no remaining HRP activity as demonstrated by the complete lack of reactive cells in Figure 5B.
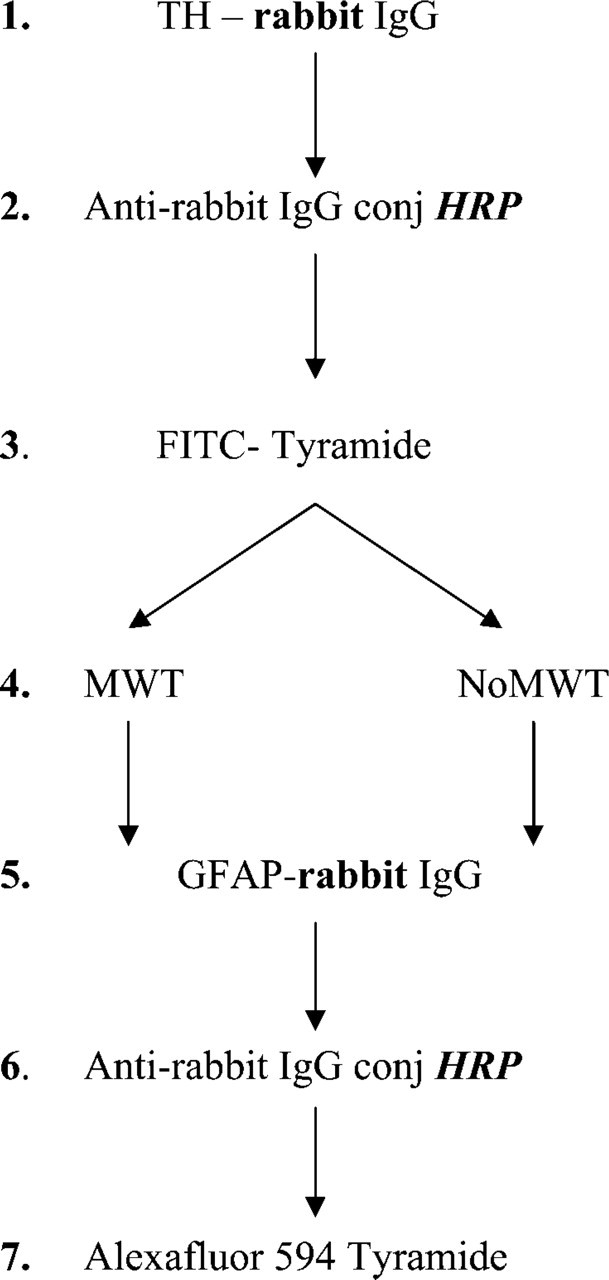
Step by step illustration of the method applied to test the effect of MWT on TH and GFAP (astrocyte-specific marker) fluorescent double immunostaining performed by using primary antibodies from the same host animals (rabbit) and amplification of the signals. We used the same HRP-conjugated anti-rabbit IgG as secondary antibody for both stainings. The potential species and peroxidase enzyme (HRP) cross-reactivity problems that might result in false colocalization signals are indicated by bold characters.
When using fluorescent secondary antibodies, we found that subsequent MWT significantly reduces the intensity of the fluorochrome conjugates. The effect of MWT on different kinds of fluorescent conjugates is shown in Figures 6A-6F. Vasopressin-associated neurophysin immunostaining was performed in mouse supraoptic nuclei using different dyes. In coronal brain sections, vasopressin-neurophysin gives a bright green signal when AlexaFluor 488-conjugated secondary anti-body is used (Figure 6A). Intensity of the fluorescent staining, however, is not preserved following MWT (Figure 6B). Staining with the same primary antibody as in Figure 6A and using biotinylated secondary antibody followed by an AlexaFluor 488-conjugated streptavidin results in a bright staining (Figure 6C) that is significantly diminished, but still clearly visible after MWT (Figure 6D). In Figure 6E, the vasopressin-neurophysin immunostaining is detected by a HRP-conjugated secondary antibody, and FITC-conjugated tyramide is then used as a substrate. In this case, the MWT (Figure 6F) does not affect the intensity of the fluorescence signal.
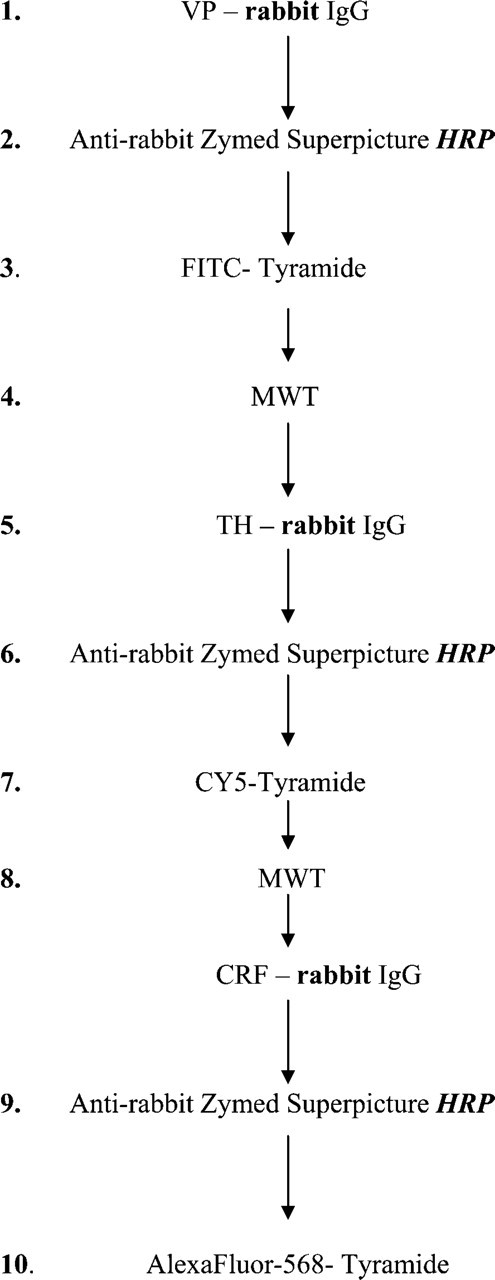
Step by step illustration of the method applied to test the effect of MWT on a fluorescent triple immunostaining performed by using primary antibodies from the same host animals (rabbit) and amplification of the signals by using the same HRP-conjugated anti-rabbit IgG as secondary antibody. The potential species and per-oxidase enzyme (HRP) cross-reactivity problems that might result in false colocalization signals are indicated by bold characters.
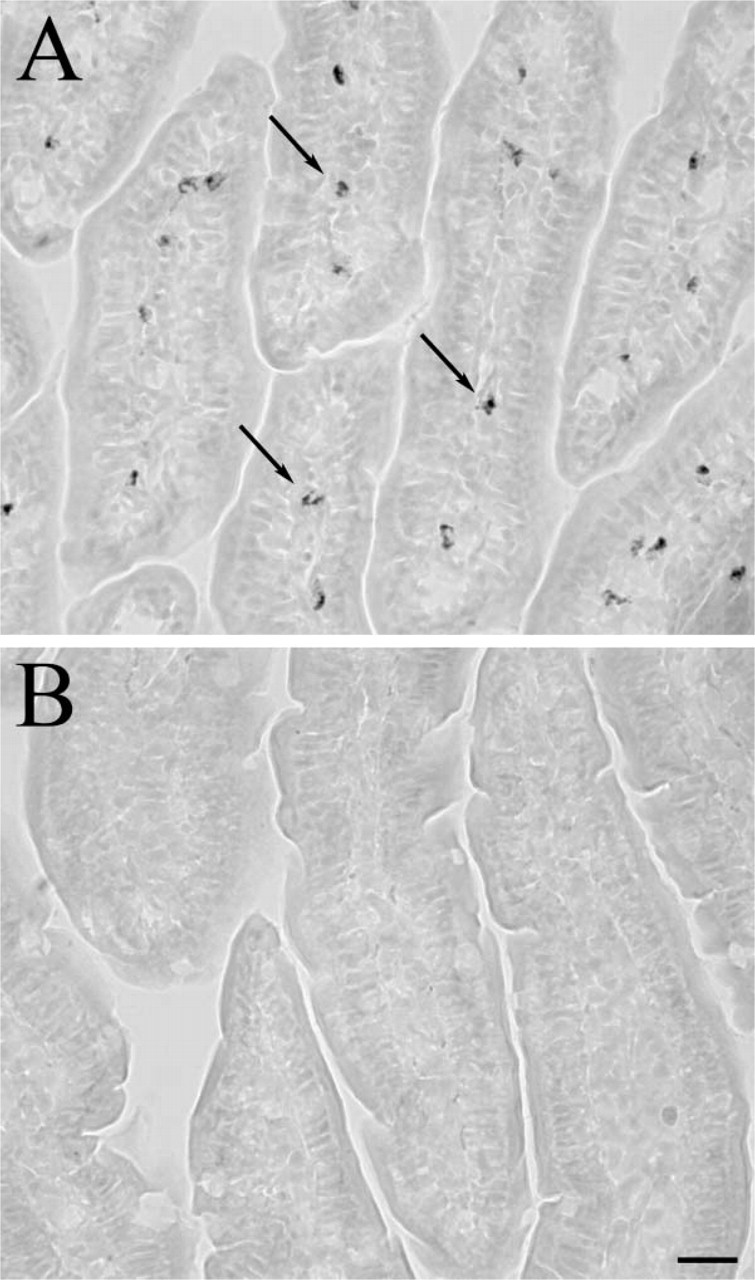
MWT eliminates endogenous peroxidase activity. (
In a double-immunostaining procedure, a coronal section across the rat striatum was first stained with a microglial marker (Iba), and the staining was developed with streptavidin AlexaFluor 488 (green). The second primary antibody was directed against TH and visualized with anti-rabbit AlexaFluor 594 (red). Both primary antibodies were raised in rabbit. As shown in Figure 7A, the anti-rabbit AlexaFluor 594 antibody recognizes both the rabbit anti-Iba and the rabbit anti-TH antibodies. If MWT is used after the Iba staining reactions are performed (Figure 7B), there is no false Iba/TH colocalization signal, and the green microglia (Iba positive) is clearly separated from the red dopaminergic (TH-positive) labeling. In our experience, swapping the order of the antibodies does not affect the staining intensity for either antigen (data not shown). Next, a double staining was performed with two primary antibodies raised in different species (Iba raised in rabbit and developed with a biotinylated secondary antibody followed by streptavidin AlexaFluor 488 and orexin raised in goat and developed using an anti-goat IgG conjugated to AlexaFluor 594). Even though the two primary antibodies were made in different species, we still saw a false double labeling of cells (arrows in Figure 7C). The false staining in this case is caused by the secondary goat antibody used for the detection of Iba, which is bound by the anti-goat AlexaFluor 594 that is meant to visualize orexin. If MWT is used after the first staining sequence, the Iba (green) and orexin (red) dyes give separate signals; thus, MWT prevents the nonspecific labeling (Figure 7D).
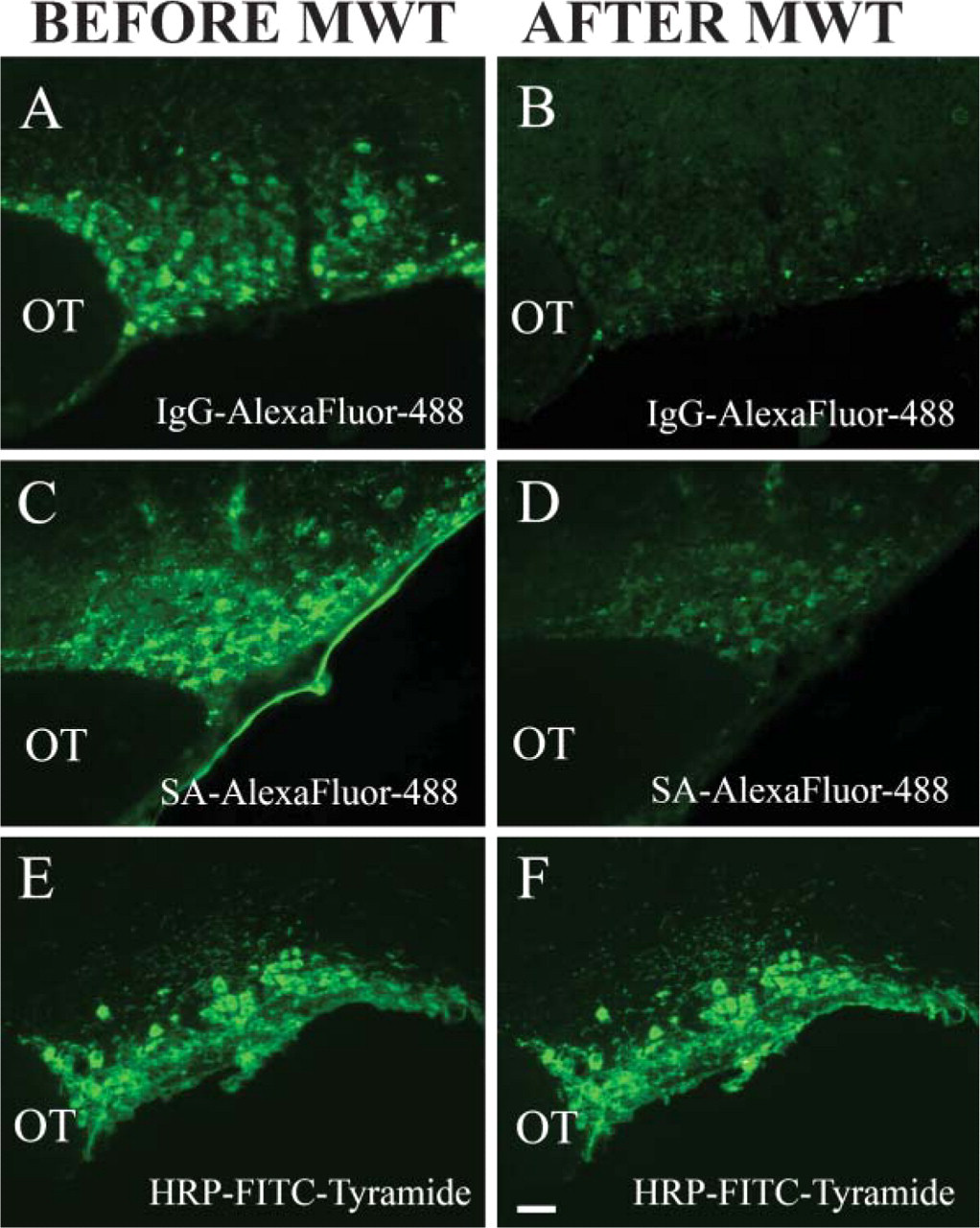
Effect of MWT on different types of fluorescent conjugates. (
In the third series of double immunostainings, we used a mouse monoclonal antibody that recognizes a neuronal-specific nuclear antigen (NeuN) and a rabbit anti-TH antibody (TH is the rate-limiting enzyme in catecholamine biosynthesis and is an accepted marker of dopamine-making cells). After the primary NeuN incubation, an anti-mouse SuperPicture was used to introduce HRP enzyme at the antigenic site. This was followed by the addition of AlexaFluor 594-Tyramide as a substrate. Next, the second primary incubation (TH) was performed followed again by the same anti-mouse SuperPicture as for the NeuN, and the reaction was finished by the addition of FITC-Tyramide as a substrate. The TH staining gives false colocalization signal with the NeuN staining (yellow cell nuclei) in Figure 8A, because the HRP enzyme that catalyzed the first tyramide reaction (red) was still capable of binding the second tyramide substrate (green) and because of the species crossreactivity. The MWT after the first sequence of reactions inactivated the HRP enzyme in the SuperPicture reagent and made it possible to perform a second consecutive peroxidase reaction without any sign of cross-reactivity. There is a nice separation of red nuclei (neurons) and green fibers (TH) in the striatum (Figure 8B). When two anti-rabbit antibodies were used (GFAP and TH) in the same section, and both stainings were amplified using different color tyramide substrates, the MWT following the first staining completely eliminated the cross-reactivity between the two detection sequences (Figure 8C). Dopaminergic neuronal fibers are shown in green (TH staining, FITC-Tyramide), and astroglia are shown in red (GFAP staining, Alexa-Fluor 594-Tyramide). Finally, to demonstrate the efficiency of the technique we performed a triple immunolabeling in the mouse hypothalamus where all three antibodies were made in rabbit, and HRP-based amplification was used three times — all in the same section and all done in 1 day. All three stainings show a distinct individual pattern characteristic to vasopressin (Figure 9A), CRF (Figure 9B), and TH (Figure 9C). There is no nonspecific staining observed when the three labels are overlaid (Figure 9D).
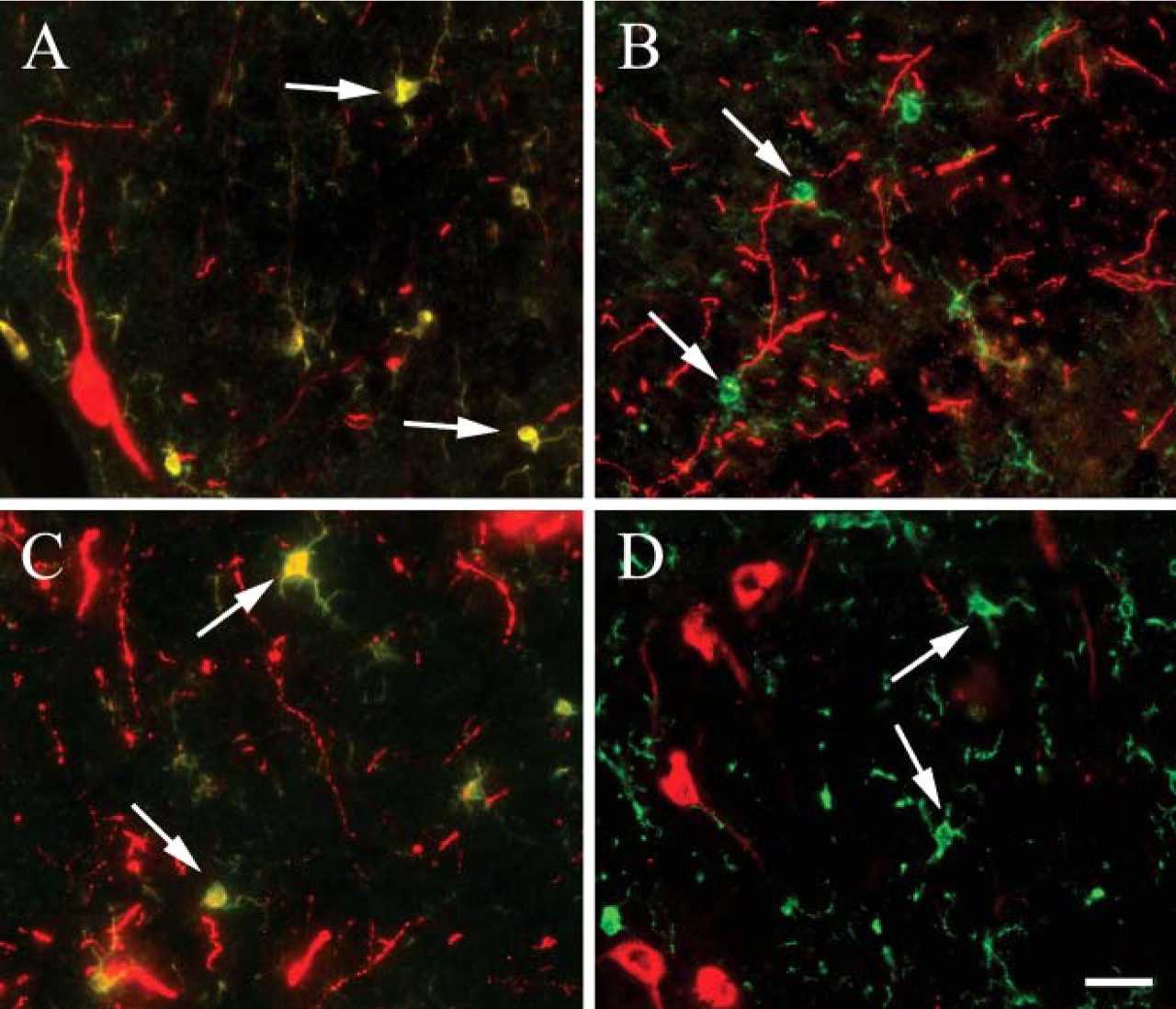
Double fluorescence immunostaining using antibodies from same host. (
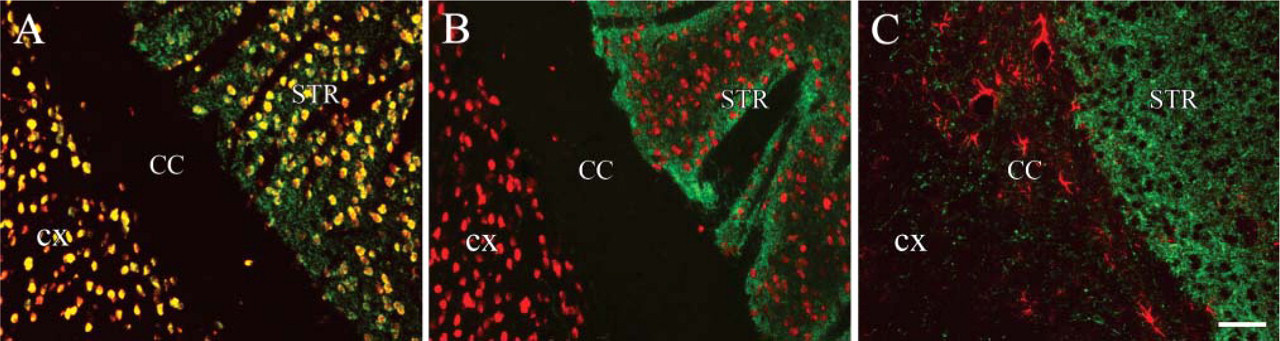
MWT for double stainings with amplification of the signals. (
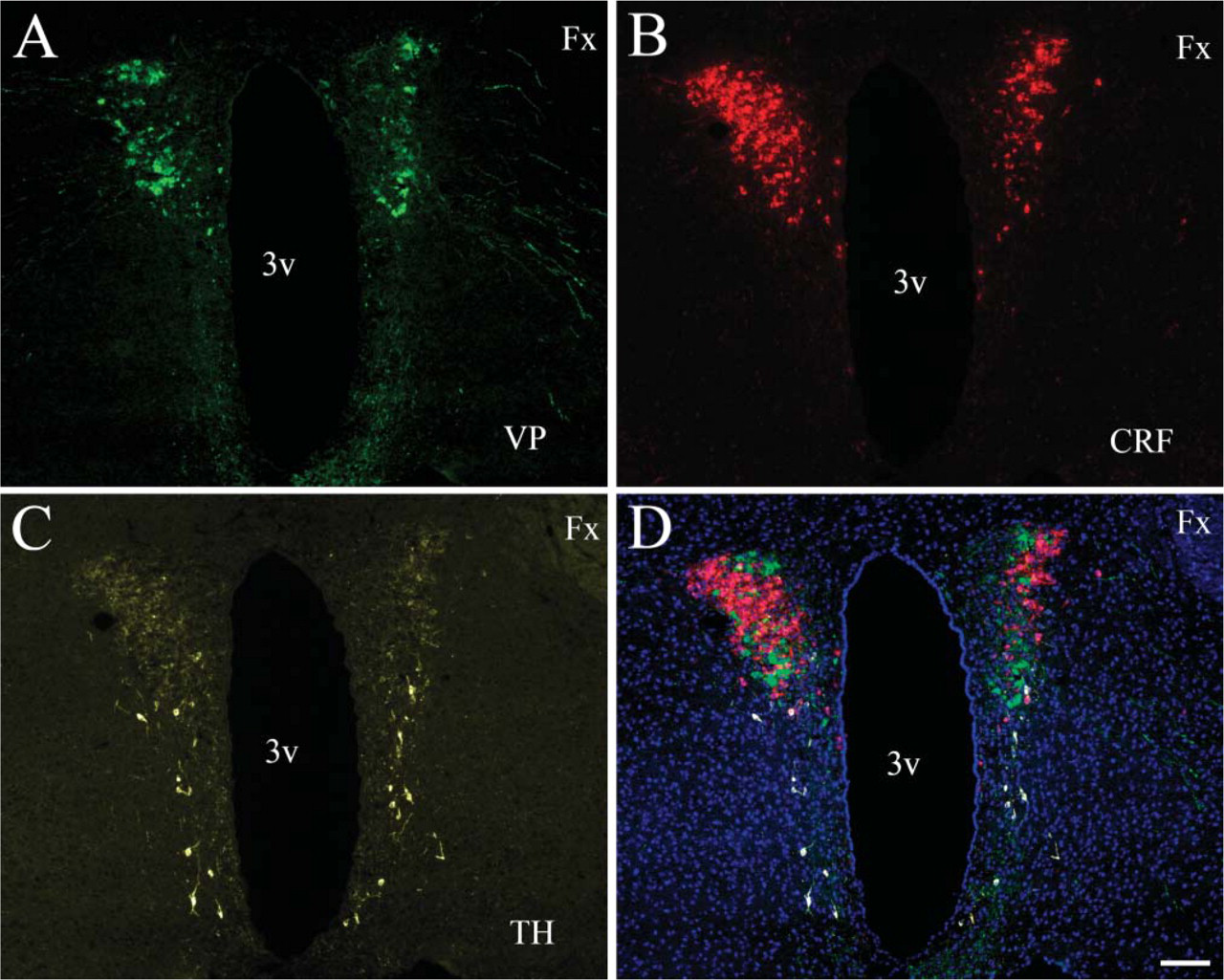
Triple immunostaining using same host antibodies. A section of the mouse hypothalamic paraventricular nucleus is shown where cells were immunostained for vasopressin (
Discussion
Use of the microwave oven as a histological tool was introduced in the early 1990s (Leong 1996; Shin et al. 2002) and involved many histological procedures such as tissue fixation, demineralization, in situ hybridization, and immunocytochemistry (Shin et al. 2002; Emerson et al. 2006; Tesch et al. 2006). Many antibodies and conditions were tested, and the major advantage of the use of microwave technology seemed to be the speed of the procedures enabling significantly faster results in human diagnostics during surgical procedures or routine laboratory work (Cuevas et al. 1994; Brown and Chirala 1995; McMahon and McQuaid 1996; Sperry et al. 1996; Munoz et al. 2004; Ridderstrale et al. 2005). A special advantage of the microwave antigen retrieval that seemed to be very useful in human pathology was the ability of using archived paraffin sections from autopsy cases. These tissues were usually very hard to work with because of the significant loss of antigenicity due to high temperatures of the paraffin-embedding procedure as well as the overfixation of these tissues by immersion in high percentage of formaldehyde (for months to years) in routine pathology (Cuevas et al. 1994; Leong 1996; Bohle et al. 1997; Bull and Harnden 1999; Relf et al. 2002; Kitayama 2005; Tesch et al. 2006). Microwave pretreatment was not only useful for immunocytochemistry but also for tissue pretreatment prior to in situ hybridization histochemistry (McMahon and McQuaid 1996; Sperry et al. 1996; Bull and Harnden 1999; Relf et al. 2002; Ridderstrale et al. 2005). We wondered if MWT could also be used to help us perform double labeling of the same sections with two (or more) antibodies that were raised in the same host. Several methods have been used to achieve this goal. The first such technique used elution of the first antibody-peroxidase complex after the water-insoluble DAB precipitate has formed (Nakane 1968). This technique includes an acid wash to remove the first antibody and is likely to cause deterioration in the quality of the tissue. Another method used immunostaining of two thin adjacent sections with the respective antibodies to try to superimpose the images to find the cells that reacted with both antibodies (Weber et al. 1982; Wolter 1985). This technique is not easy to perform and has a limitation based on section thickness and cell size (the smaller the cells the more difficult to find matching cells). Another technique was to use a secondary polyclonal monovalent F(ab) antibody after the first staining to block the excess sites from the first primary antibody. This would prevent the second secondary antibody from binding to the first primary antibody (that is from the same host). Then one can proceed to incubate with the second primary antibody followed by a secondary antibody (Negoescu et al. 1994). This technique requires very careful control experiments and is difficult and time-consuming.
Heat treatment was also suggested to denature immunoglobulins, even before MWT became available (Kolodziejczyk and Baertschi 1986). This protocol used excessive heat (130C) to remove the immunoglobulins after pictures of a staining were taken. Then a new staining was performed and new pictures were taken and compared with the first set of images. Although the technique worked reliably, there were several drawbacks. Five to seven days of tissue protection (to preserve structure in the face of excessive heat) were needed between the different stainings. Another difficulty is that it is not always easy to predict where pictures should be taken from for colocalization with a subsequent antigen. When the TSA technique became widely used in IHC, we published a protocol that utilized the sensitivity of the TSA technique to double label in the same section with two antibodies from the same host. This protocol uses a very dilute primary antibody that is not recognized by a simple secondary antibody, but can be visualized using TSA. The other primary antibody is then used at regular working dilution and is detected by a secondary antibody—without amplification (Hunyady et al. 1996). This method also needs very careful design; the optimal dilution for the first primary antibody needs to be titrated accurately and will be different for every unique antibody. Furthermore, if both antigens are rare and both need to be amplified, the technique cannot be used.
Lan et al. (1995) suggested the use of microwave to denature already bound antibody molecules to use two primary antibodies and detect nuclear and cytoplasmic antigens using alkaline phosphatase and HRP enzymatic visualization. Tornehave et al. (2000) suggested the use of microwave for double labeling and suggested that moderate microwaving does not elute antibodies that bind, but rather prevents them from binding to subsequently applied reagents. These authors suggested microwaving between the first and second staining cycles to enable double-indirect immunofluorescence staining when the antibodies used are raised in the same species. Later, however, serious questions were raised about the ability of MWT to completely abolish contaminating staining (Bauer et al. 2001). Furthermore, the loss of fluorescence due to the MWT was also not addressed in the above studies. Our protocol takes advantage of the TSA technique at several levels. First, using TSA enables detection of very rare antigens. Second, due to the very high amounts of fluorochrome binding to the peroxidase enzyme, fluorescent intensity is not affected by the MWT. Because the tyramide-fluorochrome complex is insoluble in water, once the deposit forms, the product is very stable and the tissue can be microwaved long enough to disrupt all protein bonds as well as to block endogenous and also intentional (i.e., included in the reagents that we use intentionally) peroxidase activity in the tissue. The technique is fast and easy to perform, and using the SuperPicture complex significantly increases sensitivity. The procedure is fast, can be repeated as many times as needed, and only the tyramide-fluorochrome complex will remain at the antigenic site. Several spectrally non-overlapping fluorochromes could be used in the same section for conventional or confocal fluorescent analysis. This protocol eliminates the problems with double-staining techniques used currently and due to its sensitivity can be used in fluorescent staining procedures to detect a variety of rare antigens.
Footnotes
Acknowledgements
This research was supported by the Division of Intramural Research, National Institute of Dental and Craniofacial Research, Intramural Research Programs, National Institutes of Health. Z.E.T. is supported by OTKA T-043169.