
Abstract
The liver X receptor (LXR) is a nuclear receptor that acts as a sterol sensor and metabolic regulator of cholesterol and lipid homeostasis. Using a novel LXRα-specific antibody for immunohistochemistry, we evaluated cellular expression of LXRα in fetal rat tissues. In the fetal liver, LXRα-positive macrophages appeared at 12 days and their number peaked at 18 days of gestation. In contrast, hepatocytes expressed LXRα during the later stage of gestation, suggesting the functional development of the liver during ontogeny. Later, macrophages in spleen and thymus expressed LXRα, and some mononuclear cells in the vascular lumen compatible to primitive/fetal macrophages in the fetal circulation were found to express LXRα. In vitro, rat monocytes did not express LXRα, but monocyte-derived macrophages cultured in the presence of macrophage-colony stimulating factor revealed the distinct expression of LXRα in nucleoli. These findings suggest that LXRα plays a role in the differentiation of fetal macrophages, particularly hepatic macrophages, in rat development.
L
Using a newly produced antibody, we previously demonstrated that LXRα protein was expressed in hepatocytes, adipocytes, and macrophages in human tissues as well as in foam cells in atherosclerotic lesions (Watanabe et al. 2005). In this study we examined the cellular expression of LXRα in fetal rat tissues involved in lipid metabolism, such as a yolk sac and liver and tissue macrophages. We also investigated expression of LXRα and nuclear localization in monocyte-derived macrophages in vitro.
Materials and Methods
Plasmid and Transient Transfection
Rat LXRα expression vector was constructed using cDNAs obtained from reverse transcription polymerase chain reaction (RT-PCR) of rat insulinoma INS-1E cells poly(A)+ RNA. Primers for full-length rLXRα (nucleotides 22-1363, GenBank Accession Number NM031627) were as follows: 5′-GGGGTACCGAGATGTCCTTGTGGCTG-3′ and 5′-GGGGATCCGTCATTCGTGGACATCCCAG-3′. PCR-amplified products were digested and inserted into the KpnI and BamHI site of pcDNA3 vector (Invitrogen; Carlsbad, CA). CHO cells were plated in 10 cm2 plates at 5 × 105 cells 18 hr prior to transfection. Transfections were performed with TransIT LT-1 transfection reagent (Mirus; Madison, WI) using 8 μg of pcDNA3-rLXRα expression vector per plate. Nuclear extracts for immunoblotting were prepared as described previously (Sakai et al. 1996).
Animals
Wistar rats (260–300 g) were obtained from Charles River Inc. (Tokyo, Japan) and maintained under standard conditions at the Laboratory Animal Center of Niigata University School of Medicine. All animals were allowed free access to laboratory chow and tap water. Rats were mated overnight. The day when vaginal plug formation was detected was counted as day 1. Pregnant or newborn rats were killed the same day by ether anesthesia. Fetal tissues, including the liver and yolk sac from 11 days of gestation (E11) to E20 and rat tissues at 1, 14, and 56 days after birth, were obtained for immunohistochemical (IHC) staining. The tissue sample of liver for real-time PCR was obtained from rats at E14, 16, 18, and 56 days after birth. For immunoblotting, tissue samples of heart, lung, liver, spleen, and kidney were obtained from rats at 56 days after birth. For the culture of peripheral blood mononuclear cells (PBMCs), peripheral blood from rats at 56 days after birth was also collected from the aorta.
Cell Preparation and Culture
PBMC preparation and cell culture were previously described (Liu et al. 2005). Briefly, PBMCs were isolated by Lymphoprep (Axis Shild; Oslo, Norway). PBMCs were suspended at a concentration of 1 × 106 cells/ml in RPMI 1640 medium (Sigma-Aldrich; Irvine, UK) without serum and placed in Lab-Tek multiwell tissue culture chambers (Nagle Nunc International; Naperville, IL). After a 2-hr incubation at 37C, non-adherent cells were removed and RPMI 1640 medium with 10% fetal bovine serum (FBS; JRH Biosciences, Lenexa, KS) containing 1000 U/ml of macrophage-colony stimulating factor (kindly provided by Morinaga Milk Co.; Kanagawa, Japan) was added. For IHC staining, the 2-hr incubated cells and 7-day cultured cells were washed with PBS, fixed with cold acetone for 30 min, and prepared with 0.5% Triton X-100 for 15 min. May-Giemsa staining was performed for morphological examination of cultured cells. The 2-hr incubated cells and 7-day cultured cells were collected for real-time PCR and immunoblotting.
Antibodies
Mouse anti-human monoclonal antibody LXRα (PPZ0412) was purchased from Perseus Proteomics Inc. (Tokyo, Japan) (Watanabe et al. 2005). A mouse anti-rat monocyte/macro-phage and dendritic cell monoclonal antibody ED1 was purchased from BMA Biomedicals (Augst, Switzerland). Mouse monoclonal antibody for class A macrophage scavenger receptor (MSR-A) was kindly provided by Prof. M. Takeya (Second Department of Pathology, Kumamoto University School of Medicine). Chicken monoclonal antibody for actin (JLA20) was purchased from Oncogene Sciences (Uniondale, NY). Anti-LXRα, anti-ED1, and anti-MSR-A antibodies were used for IHC at dilutions of 1:100, 1:500, and no dilution, respectively. Anti-LXRα and anti-actin antibodies were used for immunoblotting at dilutions of 1:1000 and 1:2000, respectively.
IHC
Tissues were fixed with 10% formalin and embedded in paraffin. To retrieve antigens, specimens were deparaffinized and heated in sodium citrate buffer, pH 6.0, for 15 min at 120C. Endogenous peroxidase activity was quenched using 3% H2O2-methanol for 15 min, and then the specimens were blocked with 10% normal goat serum before overnight incubation with the primary antibodies at 4C. As the second step, peroxidase (PO)-conjugated goat anti-mouse immunoglobulin (F[ab']2; Nichirei Corporation, Tokyo, Japan) was reacted for 60 min. They were carefully washed three times with PBS in each step. To visualize PO activity, 3,3′-diaminobenzidine (DAB; Dojindo Laboratories, Kumamono, Japan) was used as the substrate in 0.05 M Tris-HCl buffer (pH 7.6) containing 0.01% H2O2. Nuclear staining was performed with hematoxylin. IHC staining for the culture cells was the same as above except for retrieving antigens. IHC double staining between LXRα and ED1 was performed to investigate whether LXRα-positive cells were tissue macrophages. Briefly, the first reaction was performed using anti-LXRα antibody and the specimens were visualized as brown with DAB; then, the specimens were incubated at 4C overnight with ED1. They were visualized as blue with 4-chloro-1-naphthol. IHC staining patterns were examined using samples collected from three individual rats. The numbers of LXRα-, ED1-, and MSR-A-positive cells with nuclei per mm2 were counted using a micrometer.
Immunoblotting
Immunoblotting was performed as described previously (Saito et al. 2005). Briefly, samples were lysed in a solution containing 150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 1% Triton X-100 and 1 mM PMSF, 1 mM leupeptin, 1 mM aprotinin for 30 min on ice and centrifuged at 12,000 × g at 4C for 30 min. The extracted protein was resolved by SDS-PAGE. After electrophoresis, the protein was transferred to PVDF membrane (Amersham; Aylesbury, UK). Blots were pretreated with 5% skim milk overnight and then incubated with anti-LXRα and anti-actin for 1 hr. They were then incubated with secondary anti-mouse IgG horseradish peroxidase-conjugated antibody (Amersham) diluted 1:2000 for 30 min, washed three times with PBS between each step of the procedure, and visualized with the ECL detection system (Amersham) according to the manufacturer's instruction.
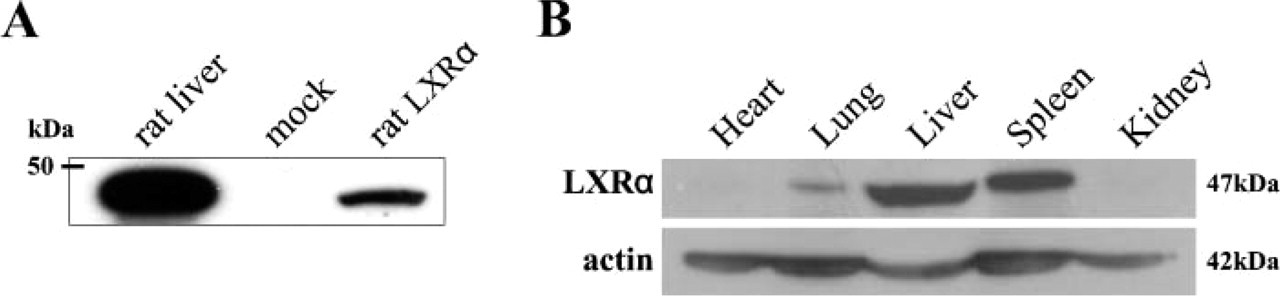
Specificity of monoclonal antibody PPZ0412 and expressions of LXRα in rat adult tissues. (A) Nuclear extracts obtained from CHO cells transfected with rat LXRα and adult rat liver showed the specific band corresponding to 47 kDa. (B) Rat LXRα protein corresponding to 47 kDa was detected in the lung, liver, and spleen. No LXRα protein was detected in the heart and kidney.
Isolation of RNA and Analysis of mRNA
Total RNA was isolated from the liver tissues and cultured cells using the method of acid guanidinium-phenolchloroform. Of the total RNA, 1 μg was mixed with 1X RT buffer (Invitrogen), 0.01 M of dithiothreitol (Invitrogen), 20 U of RNase inhibitor (Promega), 500 μM of deoxynucleoside triphoshates (dNTPs; Pharmacia, Uppsala, Sweden), 1 μg of random primer (Promega), 100 U of reverse transcriptase (Invitrogen), and distilled water to bring the final volume to 20 μl. The mixture was incubated at 42C for 60 min and then boiled at 95C for 3 min. Real-time PCR was performed using a chromo-4 four-color real-time PCR system (Bio-Rad; Hercules, CA) with SYBR Green technology, and the threshold cycle numbers were calculated using Opticon Monitor ver 3.1 (Bio-Rad). The reaction mixture consisted of 1 μl of sample cDNA, 1X iQ SYBR Green Supermix (Bio-Rad), 200 nM of each of the primers, and distilled water to bring to the final volume of 20 μl. PCR was carried out with the following primers described elsewhere: LXRα (Hoekstra et al. 2003), forward: 5′-TCAGCATCTTCTCTGCAGACCGG-3′, reverse: 5′-TCATTAGCATCCGTGGGAACA-3′; β-actin (Watanabe et al. 2003), forward: 5′-TGGAATCCTGTGGCATCCATGAAAC-3′, reverse: 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. The protocol was 40 cycles of denaturation at 95C for 10 sec, annealing at 55C for 30 sec, and elongation at 70C for 30 sec. Reactions were performed in duplicate and threshold cycle numbers were averaged. β-actin was used for normalizing expression data.
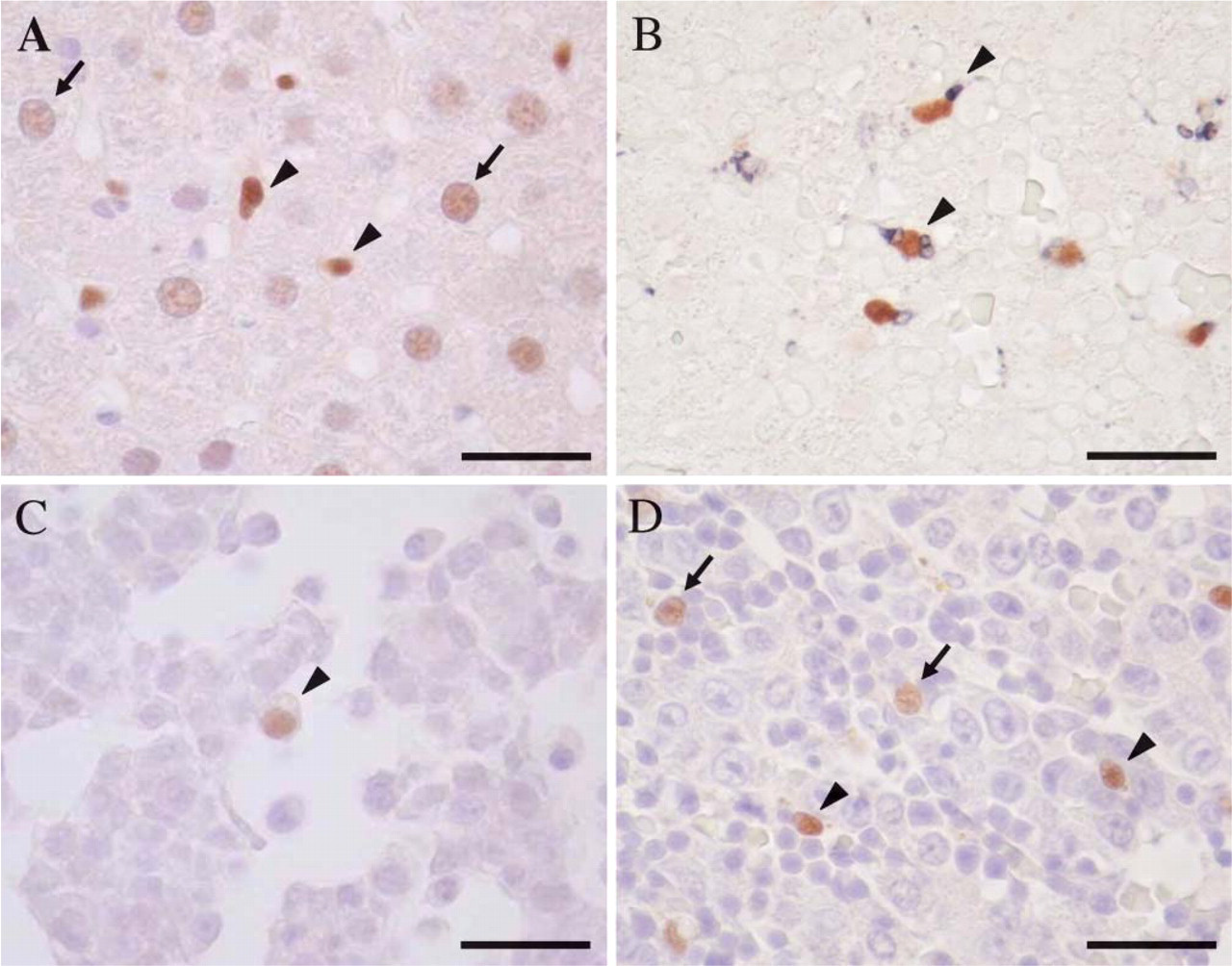
Immunohistochemical staining of LXRα in adult vs fetal rat liver. LXRα-positive cells were found as a homogeneous nuclear staining pattern. (A) LXRα-positive hepatocytes (arrow) and Kupffer cells (arrowhead) were seen in adult rat liver. (B) Immunohistochemical double staining demonstrated that LXRα (brown) and ED-1 (blue) double-positive cells were seen in more than half of the ED1-positive macrophages (arrowhead) at E18. (C) LXRα-positive macrophages (arrowhead) appeared in the fetal liver at E12. (
Immunofluorescence
Double immunofluorescence was described previously (Iguchi et al. 2005). Briefly, cells cultured for 7 days were washed with PBS, fixed with 4% paraformaldehyde for 5 min at 4C, prepared with 0.5% Triton X-100 for 5 min at 4C, and blocked with 10% goat serum for 30 min at room temperature. They were stained with an anti-nucleophosmin (Zymed; South San Francisco, CA) and an anti-LXRα labeled with Zenon Alexa Fluor 488 labeling kit (Molecular Probes; Eugene, OR) at dilutions of 1:100 and 1:50, respectively. For nucleophosmin detection, Alexa Fluor 594 goat anti-mouse IgG (Molecular Probes) was used as a secondary antibody at a dilution of 1:2000. Control experiments were carried out by omitting the primary antibody. Specimens were counterstained with DAPI (Vector Laboratories; Burlingame, CA). Immunofluorescence was captured with a confocal laser-scanning microscope (LSM510META; Carl Zeiss, Jena, Germany).
Statistics
Three rats were examined for each procedure. Real-time PCR was carried out in duplicate. Data were expressed as the mean ± SD. Statistical analysis was carried out with SPSS version 11.0 (SPSS, Inc.; Chicago, IL). Correlation test between continuous quantitative and qualitative variables (t-test) was used to determine significances; p<0.05 was considered statistically significant.
Results
LXRα-specific Antibody for Rat Tissues
Figure 1A indicates the results of immunoblotting with PPZ0412 using protein obtained from CHO cells transfected with the rat LXRα expression vector. The antibody bound specifically to the rat LXRα protein with an apparent molecular mass of 47 kDa. Amino acid sequences of human LXRα protein are highly similar to rat LXRα, which has higher homology than human LXRβ. The antibody recognized the human LXRα ligand-binding domain (LBD), and the amino acid sequence is almost the same. The antibody did not cross-react with the rat LXRβ protein (data not shown). These findings suggested that the antibody is specific for rat LXRα protein.
Immunoblotting revealed a distinct band corresponding to 47 kDa in the tissue samples of lung, liver, and spleen taken from adult rats. No distinct band was detected in the heart and kidney (Figure 1B). Immunohistochemically, expression of LXRα in adult rat tissue was clearly found in the nuclei of hepatocytes (Figure 2A), Kupffer cells, alveolar macrophages, and macrophages in the spleen (Table 1). Macrophages were confirmed by the IHC double staining using anti-LXRα and anti-ED1 antibodies (Figure 2B). LXRα staining pattern was homogeneous in the nuclei. Kupffer cells showed higher intensity of LXRα expression than hepatocytes (Figure 2A). LXRα expression pattern of adult rat tissues was the same as that of human tissues, indicating that this antibody is available for IHC staining of rat tissues.
LXRα-expressing cells in fetal, newborn, and adult rat tissues
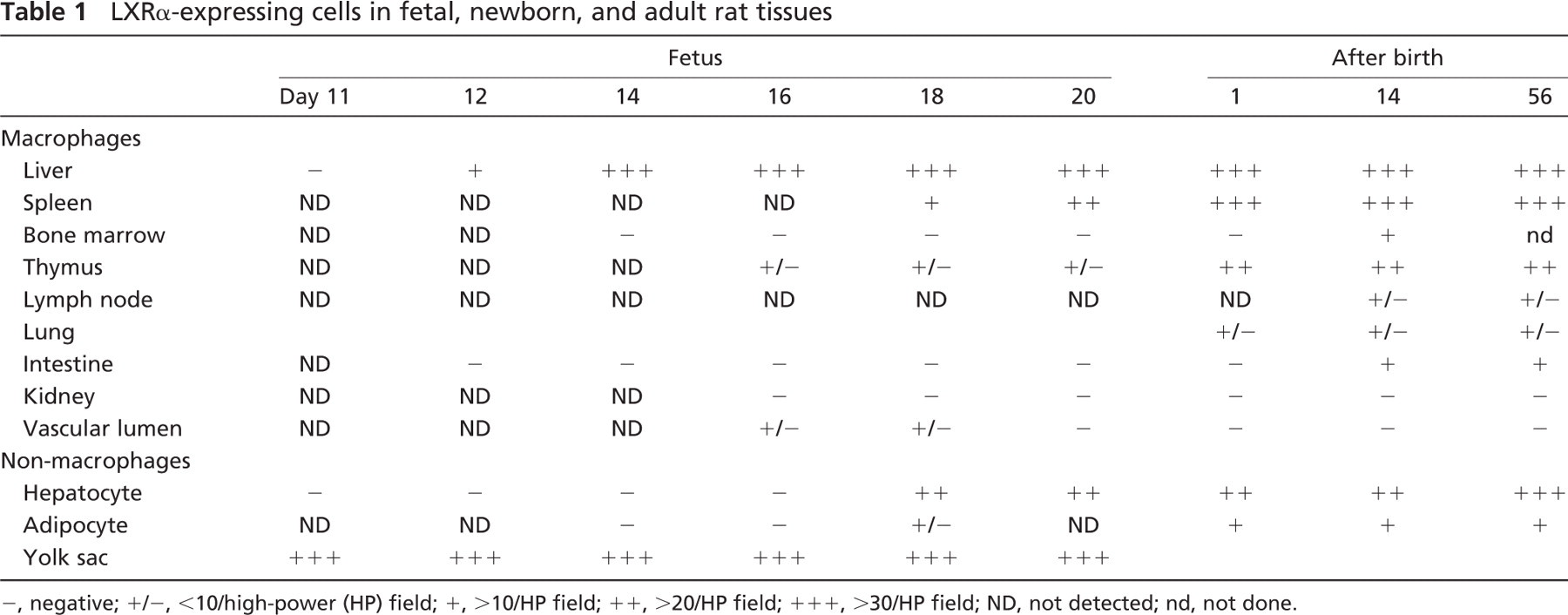
−, negative; +/−, <10/high-power (HP) field; +,>10/HP field; ++,>20/HP field; +++, >30/HP field; ND, not detected; nd, not done.
Expression of LXRα in Fetal Macrophages and Other Cells
LXRα expression was first detected in the endodermal layer of the yolk sac at E11, and LXRα expression in the yolk sac was observed throughout the fetal period (Table 1). However, no LXRα-positive cells were present in the stroma or in the vascular lumen of the yolk sac.
LXRα-positive macrophages first appeared in the fetal liver at E12 (Figure 2C) (Table 1). LXRα-positive hepatic macrophages increased with gestational days (Figure 2D). We compared expression of LXRα, ED1, and MSR-A in hepatic macrophages (Figure 3A). LXRα-and ED1-positive cells showed a peak at E18 and E16, respectively. MSR-A-positive cells increased with time and were highest in the adult liver.
Hepatocytes were negative in LXRα immunostaining until E16. LXRα-positive hepatocytes appeared at E18 (Figure 2D) (Table 1). Nuclear staining of hepatocytes became prominent after birth, but the intensity of the nuclear staining of hepatocytes was much weaker than that of macrophages (Figures 2A and 2D). LXRα mRNA level of the fetal liver was highest at E18 (Figure 3B). However, LXRα mRNA level of the adult liver was much higher than that of the fetal liver because of the increased expression of LXRα mRNA in hepatocytes.
Interestingly, LXRα-positive mononuclear cells were found in the vascular lumen at E16 and E18 (Figure 4A). In contrast, no LXRα-positive mononuclear cells were detected in the vascular lumen after birth (Figure 4B). We have reported that primitive/fetal macrophages circulate in the peripheral blood in the early stage of the fetal period, whereas monocytes first appear in blood at late gestational days and increase with each fetal day (Takahashi et al. 1996). These findings suggested that the LXRα-positive cells may be fetal macrophages in the fetal circulation.
At 16 or 18 days of gestation, LXRα-positive macrophages were detected in the spleen and thymus (Table 1). Weakly positive expression of LXRα was observed in the brown adipose tissue at 18 days of gestation (data not shown). After birth, LXRα-positive macrophages were present in several tissues (Table 1) as reported previously (Watanabe et al. 2005).
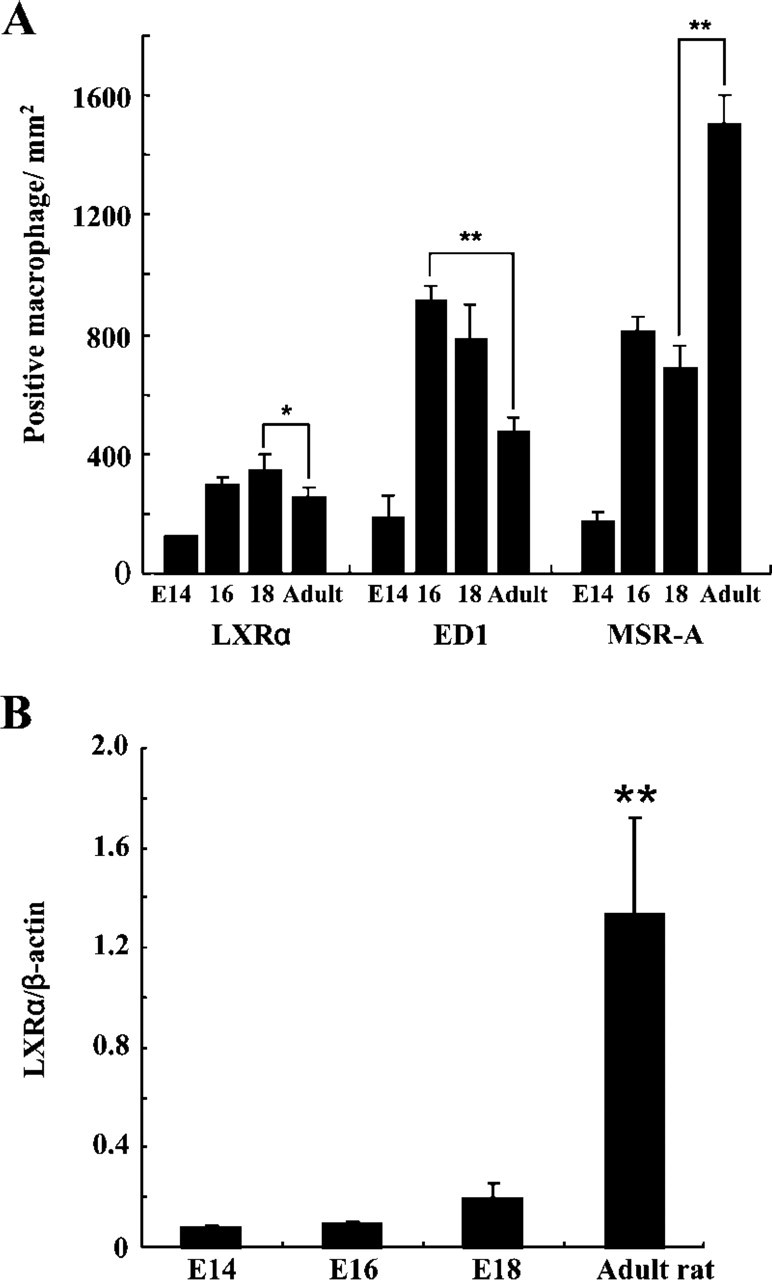
Number of LXRα-expressing macrophages and mRNA level of LXRα in adult vs fetal rat liver. (A) The number of LXRα-and ED1-positive cells showed a peak at E18 and E16, respectively. MSR-A positive cells increased with time and were highest in adult liver. Data are expressed as the mean ± SD. The numbers of LXRα-positive cells at E18 and ED1-positive cells at E16 were significantly more than in adult rat, respectively. The number of MSR-A-positive cells in adult rat was significantly higher than at E18. (B) Quantitative mRNA level of LXRα was the highest at E18 in the fetal liver. Quantitative mRNA level of LXRα after birth was significantly higher than in fetal liver. ∗ p<0.05, ∗∗ p<0.01.
Expression of LXRα in Monocyte-derived Macrophages
To investigate the relationship between expression of LXRα and macrophage differentiation, we cultured monocytes obtained from adult rats. Adherent PBMC monocytes incubated for 2 hr were small, round in shape, and with scant cytoplasm (Figure 5A). They showed negative expression for LXRα (Figure 5B) and MSR-A (Figure 5C). In contrast, cells cultured for 7 days were large and with abundant cytoplasm (Figure 5D). Most of them distinctly expressed both LXRα (Figures 5E and 6A) and MSR-A (Figure 5F). LXRα expression was observed in nuclei as dots and MSR-A expression was predominantly observed on the cell membrane in these cells. LXRα mRNA and protein levels were also paralleled with LXRα expression in these cells (Figures 6B and 6C).
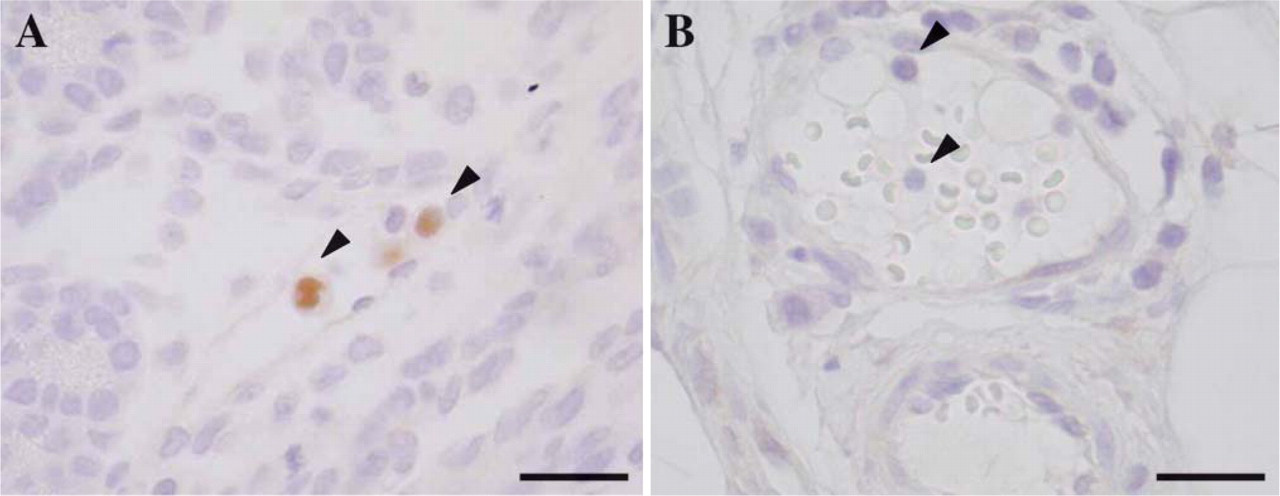
Immunohistochemical staining of LXRα in mononuclear cells in adult vs fetal rat vascular lumen. (A) LXRα-positive mononuclear cells (arrowhead) were seen in the vascular lumen at E18. (B) No LXRα-positive mononuclear cells (arrowhead) were detected in the vascular lumen after birth. Bar = 40 μm.
Interestingly, immunofluorescence double staining showed a colocalization of LXRα and nucleophosmin (Figure 7). Nucleophosmin, also known as numatrin, No38 and B23, is characterized as a nucleolar protein functioning in the processing and transport of rRNA (Borer et al. 1989). These findings suggested that LXRα is localized in nucleoli.
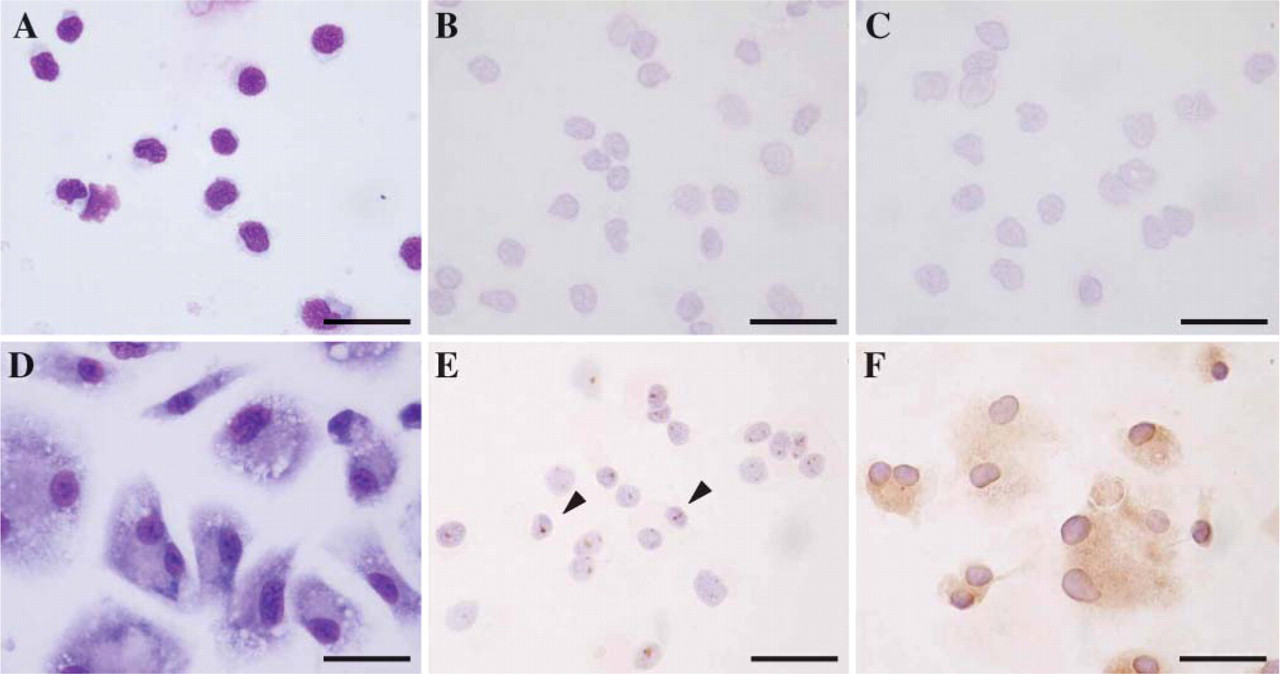
Morphology and immunohistochemical staining of LXRα in rat monocyte-derived macrophages. (A) Monocytes were small, round in shape, and with scant cytoplasm. Expressions of LXRα (B) and MSR-A (C) were not seen in 2-hr incubated monocytes. (D) Monocyte-derived macrophages after 7-day cultures were large and had abundant cytoplasm. Almost all macrophages were positive for LXRα (E) and MSR-A (F). LXRα expression was observed in nuclei as dots (arrowhead), and MSR-A expression was predominantly observed on the cell membrane in these cells. Bar = 40 μm.
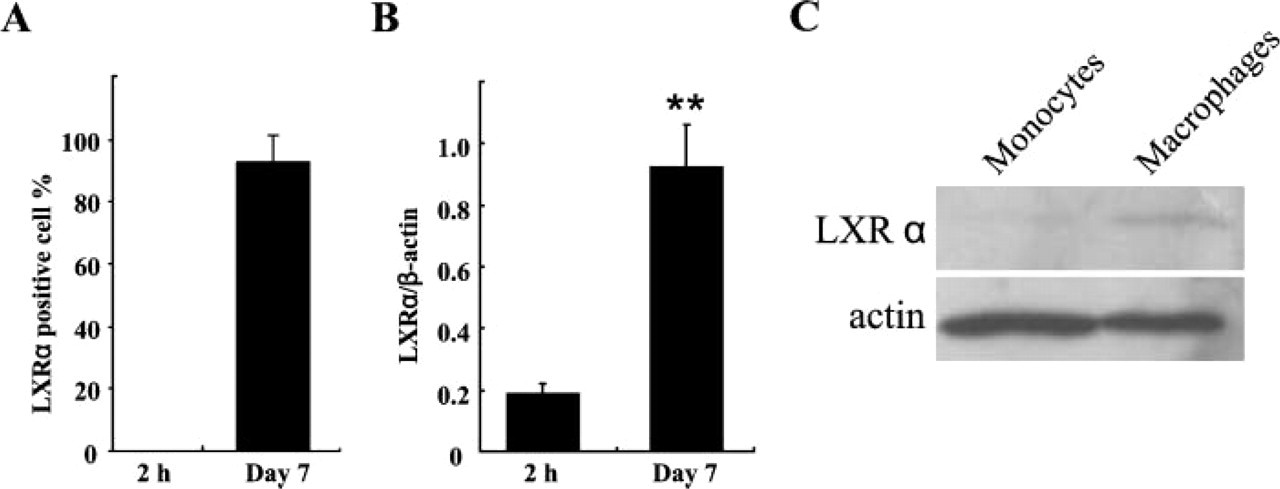
The number of LXRα-expressing macrophages and mRNA and protein level of LXRα in rat monocyte-derived macrophages. (A) Most of the monocyte-derived macrophages were positive for LXRα, whereas monocytes showed no positive cells for LXRα. Data are expressed as the mean ± SD. (B) Quantitative mRNA level of LXRα clearly increased in monocytederived macrophages (7-day cultures). ∗∗ p<0.01. (C) LXRα proteins corresponding to 47 kDa were detected in monocyte-derived macrophages (7-day cultures), whereas a faint band was detected in monocytes.
Discussion
Using a newly produced antibody, the present study demonstrated that LXRα protein was detected in macrophage lineage cells as well as in endodermal cells of the yolk sac, hepatocytes, and adipose tissue during rat development.
Among the macrophage lineage cells, LXRα protein expression was first confirmed in hepatic macrophages during rat development, followed by appearance of LXRα-positive macrophages in other tissues. The number of LXRα-positive hepatic macrophages showed a peak just before birth, and LXRα mRNA levels in fetal liver were highest just before birth. Macrophages are abundant in fetal liver and actively phagocytize various blood cells (Naito et al. 1997), suggesting that hepatic macrophages may be most active in lipid metabolism or other functions among fetal tissue macrophages.
Kohro et al. (2000) reported that LXRα mRNA is the most highly induced transcriptional regulator during differentiation from human primary culture mono-cytes to macrophages. We have shown that mRNA and protein expression of LXRα in rat macrophages was much higher than those in monocytes in vitro. These results provide evidence that LXRα expression is regulated by the differentiation and functional status of macrophages. Rat monocytes did not express LXRα by IHC, whereas we have found LXRα-expressing mono-nuclear cells in the vascular lumen of rat fetuses. Immature macrophages, called primitive macrophages, first appear in the yolk sac and differentiate into fetal macrophages (Takahashi et al. 1996; Naito et al. 1997; Watanabe et al. 2003). Although there were no LXRα-positive cells in the yolk sac, LXRα-positive cells were found in the vascular lumen of rat fetuses at E16 and E18. IHC staining also demonstrated not only LXRα-positive cells, but also ED1-positive cells in the vessels (data not shown). These findings suggest that LXRα-positive cells in the fetal circulation are not monocytes, but fetal macrophages.
The distribution of LXRα in the nucleus was reported in the LXRα-overexpressing cells (Morello et al. 2005). To define the intranuclear localization of LXRα more precisely, we examined rat monocyte-derived macrophages by immunofluorescence microscopy. Although the nuclear staining pattern of LXRα was homogeneous in tissue sections (Watanabe et al. 2005), a dot-like staining pattern was observed in cultured macrophages. Immunofluorescence double staining revealed the colocalization of LXRα and nucleophosmin, the latter of which is identified as a nucleolar protein (Borer et al. 1989). It is known that nucleolar size affects the rapid cell proliferation in cancer cells (Derenzini et al. 2000). Lipopolysaccharide-stimulated macrophages revealed upregulation of nucleophosmin and prevented apoptosis (Zhang et al. 2006). Regulation of the PPAR-γ gene through LXRα is recognized to control cellular differentiation and apoptosis in a human promyelocytic cell line, HL-60 (Kaul and Anand 2003). These findings suggest that the intranucleolar localization of LXRα is transcriptionally significant in macrophage differentiation.
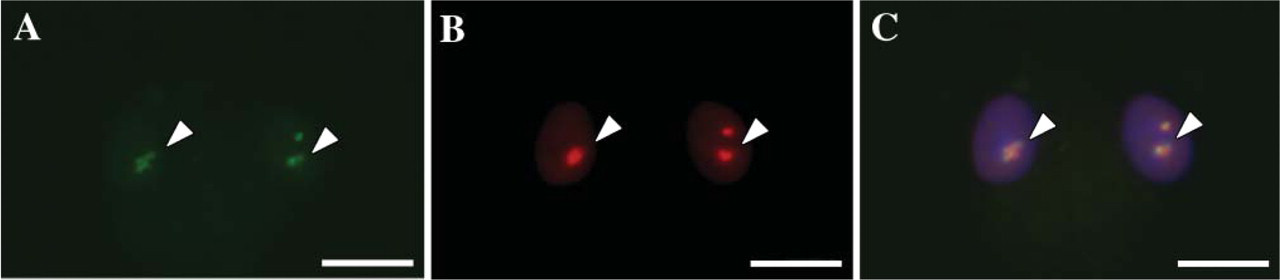
Double immunofluoresence of rat monocyte-derived macrophages. LXRα and nucleophosmin colocalized in the nucleoli of monocyte-derived macrophage. (A) LXRα (arrowhead). (B) Nucleophosmin (arrowhead). (C) Merged image with DAPI nuclear staining (arrowhead). Bar = 20 μm.
The liver plays a significant role in lipid metabolism, and hepatocytes in adult animals are the major cell type expressing LXRα. However, expresssion of LXRα in hepatic macrophages preceeded that in hepatocytes in the fetal liver. It is reported that liver acyl-CoA cholesterol acyltransferase activity is low in the early stage of gestation (Ding and Lilburn 2000). LXRα is involved in the regulation of genes contributing to hepatobiliary cholesterol and bile acid homeostasis, but nuclear receptors involved in bile formation are poorly developed during the fetal stage (Balasubramaniyan et al. 2005). This biochemical evidence coincides with LXRα expression in hepatocytes in the late stage of the rat fetal period. LXRα expression was first detected in endodermal cells of the yolk sac at E11. Because the yolk sac mediates the transfer of lipid from the yolk to the circulation (Ding and Lilburn 2000), LXRα expression in the yolk sac may play a crucial role in lipid transport before the development of liver function.
In summary, using IHC and immunoblotting techniques we clarified LXRα expression in fetal macrophages. In addition to lipid metabolism, LXRα may play an important role in the differentiation of fetal macrophages, particularly hepatic macrophages.
Footnotes
Acknowledgements
This study was supported by the Program of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation, the Focus 21 project of the New Energy and Industrial Technology Development Organization, and the Special Coordination Fund for Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology (Japan).
We thank S. Momozaki, K. Ohyachi, and T. Aoyama (Division of Cellular and Molecular Pathology, Department of Cellular Function, Niigata University Graduate School of Medical and Dental Science) for their excellent technical assistance.