
Abstract
Western blotting is one powerful research method to specifically detect proteins. However, it has been barely possible to investigate microscopic volumes of tissue so far because of the required minimum volumes and the pretreatment. Herein, we describe a method of performing Western blots directly from the histological section of frozen or paraffin-embedded tissue. Small histological areas of a mouse brain were lysed by section lysis buffer, subjected to a miniaturized SDS-PAGE, and detected by immunoblotting. Thereby, an area equivalent to only 15 cortical neurons of mouse cortex was detectable. This offers the possibility of correlating histological findings to biochemical investigations. In addition, enzymatic pretreatment was applied to identify the glycosylation of the major cleavage product of the prion protein. Moreover, the section lysis buffer is a sophisticated method to conserve and investigate phosphorylation sites as demonstrated here by phopsphorylated Akt and ERK. The presented technique combines histology with Western blotting techniques and will be of value for investigations of discrete tissue areas.
R
During the past years, there have been numerous further developments regarding the indirect immunodetection by enzyme-linked or fluorescent staining methods (Gingrich et al. 2000) as well as specific ways of preparing tissue to extract protein fractions (Huttner et al. 1983) or keeping phosphorylation sites (D'Onofrio et al. 1994; Schaeffer et al. 1998). Here, pretreatment is an important step for the separation of proteins due to their dissociation from the tissue by homogenization and structural modification such as denaturation.
For example, solubilization of cell membranes with Triton X-100 increases with temperature and can also be used to separate the detergent-insoluble and highly cholesterol-enriched membrane complexes (DICs), which are functionally important platforms of cell signaling (Aplin et al. 1998; Muller et al. 2001), by ultra-centrifugation. Activities of cell signaling kinases are mostly represented by phosphorylation sites, which are rapidly inactivated by the release of endogenous phosphatases (Zippel et al. 1989). Therefore, the conservation of phophorylation requires quick handling, cold temperatures, and special inhibitors of phosphatases (Vassallo et al. 2005).
On the other hand, specific digestion prior to electrophoresis is used to unmask selective properties of proteins. For example, the misfolded and infectious isoform of the prion protein is detectable by its partial resistance to a digestion with protease that degrades the normal cellular prion protein (Prusiner et al. 1981). Physiologically, there is also a cleavage of the normal cellular prion protein, the fragments of which are unmasked in Western blot by cutting the variable sugar side chains by PNGase F (Mange et al. 2004). All these extensive pretreatments usually require large quantities of tissue.
Microscopy of neuroanatomical structures or pathological findings of the brain on histological sections are of greatest importance. However, it is barely possible to investigate such microscopic material by Western blotting and to apply elaborate techniques of preparation and processing. Although there have been recent advances of small tissue removal by biopsy of mouse brain slices, the direct correlation to histological sections is still lacking (Barr et al. 2004). Herein, we report a method of collecting and processing tissue directly from selected areas of histological sections and analyzing the respective protein expression by Western blotting with a highly sensitive immunoblotting technique. Even tiny histological areas are detected, and it is possible to apply proteolytic digestion as well as to conserve phosphorylation sites to determine kinase activities. The method is a powerful tool for the immediate combination of the histological and immunohistochemical examinations of microscopic areas of histological sections with Western blotting techniques.
Material and Methods
Histological Sections
C57/BL6-129/sw male mice were kept under standard diurnal conditions and allowed access to food and water ad libitum in accordance with animal protection standards. Mice were sacrificed (declared to the government of Bavaria, Germany), and brains were quickly removed.
For cryosections, brains were immediately frozen in liquid nitrogen. Twenty-μm-thick coronal sections were taken with Cryostat (HM 360; Microm, Waldorf, Germany) from regions at bregma and were collected on prelabeled glass slides at −12C (SuperFrost Plus; Menzel, Braunschweig, Germany).
The freshly removed brains intended for paraffin-embedded tissue were cut in pieces of ∼3 mm and subjected to ice-cold fixation solution HOPE I and after 48 hr to acetone/HOPE II over 2 hr (DCS; Hamburg, Germany) as recently published (Uhlig et al. 2004). The fixation HOPE (HEPES-glutamic acid buffer mediated organic solvent protection effect) is based on a non-cross-linking conservation of tissue by a hyperosmolar solution enriched with amino acids and a consecutive change to organic solvent acetone precipitating. Following fixation, tissue was processed from acetone to warmed paraffin infiltrating overnight. Ten-μm sections were cut from cooled paraffin block, dried at 55C for 1 hr, and deparaffinized in xylol and graded ethanol.
Cell Lines and Cell Culture
Adherent neurons from hippocampal cell line (Kuwahara et al. 1999) were maintained in six-well plates (Nunc; Wiesbaden, Germany) with Dulbecco's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum, 2 mM glutamine, 200 U/ml penicillin, and 20 μg/ml streptomycin. Prior to stimulation, cells were serum starved with DMEM and supplemented with 0.5% heat-inactivated fetal calf serum. For stimulation, 0.013 IU/ml insulin (Insuman Rapid; Aventis Pharma, Frankfurt, Germany) was added to the medium, and cells were incubated for 10 min at 37C.
Tissue Collection
Frozen or dewaxed sections were Nissl stained with cresyl violet, dehydrated in graded ethanol, and air dried, which easily allows determination of different regions of the brain. Under a stereoscopic microscope, thin borderlines were cut with a scalpel to mark and separate a region of the brain. The delineated area was overlaid with section lysis buffer (2% SDS, 0.05 M DTT, 10% glycerol, 1 mM EDTA, 60 mM Tris-HCl, pH 7.2) using a pipette. To gain the complete tissue area this process was performed twice with an initial collection step and a secondary washing step using a total volume of 20 μl/10 mm2 of section lysis buffer. This was applied to various areas, which were lysed and collected consecutively commencing at the exterior. To avoid a possible loss of volume by air drying, the entire procedure was performed on a cooled plate at the lowest temperature at which the glass slide will not fog (∼15C).
The amount of loss of section lysis buffer by air drying at room temperature was assessed by measuring the weight on a precision balance (CP224S; Sartorius AG, Göttingen, Germany).
Solubilized tissue was collected and transferred into small tubes and heated in a PCR cycler (DNA thermal cycler; Perkin-Elmer, Norwalk, CT) for 10 min at 95C. For the deglycosylation experiments, a digestion with PNGase was performed according to standard protocols of the manufacturer (New England BioLabs; Frankfurt, Germany). The reaction was stopped by boiling in the PCR cycler for 10 min at 95C.
Collection of subconfluent cell cultures (1.06 cells per well) was carried out by discarding the medium and overlaying 300 μl/well of either Triton-based buffer [150 mM NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadat, 1 mM EDTA, 1 mM EGTA, 20 mM Tris, pH 7.5, one tablet protease inhibitor (Complete; Roche, Grenzach-Wyhlen, Germany) per 10 ml] or section lysis buffer. A short centrifugation step (16,000 × g, 10 min) followed to remove any cell debris. Aliquots of 20 μl (∼67,000 cells) were boiled after adding 5 μl Laemmli buffer (Laemmli 1970) for 10 min and subjected to Western blotting.
Western Blotting and Immunodetection
Samples were subjected to Western blotting in volume as described in Results. Proteins were separated on SDS page (NuPage Novex Bis-Tris Gel 12%, 1.0 mm, 10 wells; Invitrogen, Karlsruhe, Germany) with 100 V (Power Pac 1000; Bio-Rad Laboratories, Munich, Germany) for 24 min (∼2.5 cm) in running buffer (0.1% SDS, 1 mM EDTA, 50 mM 3-(N-morpholino)propanesulfonicacid, 50 mM Tris, pH 7.2). Afterwards, gels were transferred to a polyvinyl difluoride membrane (PVDF, 0.45 μm; Immobilon, Millipore, Eschborn, Germany) by semi-dry blotting technique (Semi Phor; Hoefer Scientific Instruments, San Francisco, CA) with 68 mA/50 cm2 for 2.5 hr with blotting buffer (20% methanol, 192 mM glycine, 25 mM Tris, pH 8.3). Subsequently, the PVDF membranes were blocked in blocking buffer (0.05 g casein (I-Block; Tropix, Bedford, MA), 0.5% Tween (Fluka Chemie GmbH; Buchs, Switzerland) in PBS, pH 7.4, or Tris-buffered saline, pH 7.4, in case of detection of phospho-Akt and phospho-ERK) for 1 hr at room temperature. The respective primary antibody was diluted in the relevant blocking buffer to a concentration of 1:5000 (β-actin, 6H4, GFAP, synaptophysin, S-100, phospho-Akt) and incubated overnight at 4C with gentle agitation. The β-actin antibody was obtained from Santa Cruz (Actin I-19; Santa Cruz Biotechnology, Heidelberg, Germany), the PrP antibody 6H4 (Prionics AG; Schlieren, Switzerland), the antibodies directed against GFAP, synaptophysin, and S-100 from Dako (DakoCytomation; Hamburg, Germany), the phospho-Akt antibody and the phospho-ERK 1,2 antibody (New England BioLabs). After that, three washing steps of 15 min each at room temperature followed in blocking buffer. Bound antibodies were labeled using a secondary antibody coupled to alkaline phosphatase (polyclonal goat anti-mouse and anti-rabbit immunoglobulin/ AP; DakoCytomation) in a concentration of 1:2000 in blocking buffer. After incubation for 45 min at room temperature, three further washing steps of 15 min each at room temperature in blocking buffer followed. The PVDF membrane was then incubated with alkaline phosphatase buffer (100 mM Tris, pH 9.5). The chromogene nitroblue tetrazolium salt with 5-bromo-4-chloro-3-indolylphosphate was used in 0.4 mM concentration to generate a blue staining in up to 2 hr (Leary et al. 1983)
Digital Imaging
To calculate the area in the histological sections, the software analySIS (Soft Imaging System GmbH; Münster, Germany) was used in combination with a CCD-camera mounted on a microscope (Axiovert 200M and Axiocam MRc5; Carl Zeiss AG, Göttingen, Germany). Confocal laser microscopy (Leica, TCS System SP2; Wetzlar, Germany) was used to control the thickness of the sections. For quantification of band intensities, Western blots were scanned with a high-resolution flatbed scanner, and the density of bands was determined using Total Lab V 2.01 software (Nonlinear Dynamics; Newcastle-upon-Tyne, UK).
Results
Evaluation of Tissue Extraction from Cryosections and Paraffin-embedded Sections
Tissue collection from histological sections was investigated first. In a Nissl-stained cryosection, an area of 10 mm2 was lysed by adding 20 μl of section lysis buffer as described above. The weight before and directly after adding 20 μl of lysis buffer was measured, resulting in an increase of ∼20.4 mg (high density of glycerol). Subsequently, the section was exposed to air at room temperature for 3 min leading to a weight loss of 0.9 μg (4%), although due to the solubilization of the tissue it would be possible to collect the tissue earlier. The lysed tissue was then drawn with a pipette, resulting in a remnant of 0.4 μg on the glass slide that represents a tissue collection of ∼98% completeness at room temperature. Samples were subjected to SDS-PAGE. A clear band was detected in the Western blot using a β-actin antibody at 42 kDa.
Consequently, it was determined that the minimum area was still well detectable in Western blot. For this purpose, the area of a quarter of a coronal mouse brain section with 20 μm thickness (controlled by confocal laser microscopy) was quantified by digital imaging software and lysed with 2 μl/mm2. The collected sample was diluted several times by repeating a 1:1 dilution with section lysis buffer, resulting in a logarithmic series. Western blots stained for β-actin, glial fibrillary acidic protein (GFAP), and synaptophysin are shown in Figure 1. The corresponding size of the area of the coronal mouse brain section is calculated and given as additional information with the subheading. In 0.021 mm2, equivalent to a tissue volume of 42 nl, β-actin was instantly recognizable. The corresponding staining of GFAP and synaptophysin was somewhat weaker but still detectable. Due to the enzymatic staining, by which the alkaline phosphatase is used up through forming of insoluble formazan products, the relation between the concentrations and the densities of the bands was fitted by a non-linear regression. A suitable correlation was found here with an exponential fit following (r2 = 0.98 for β-actin):
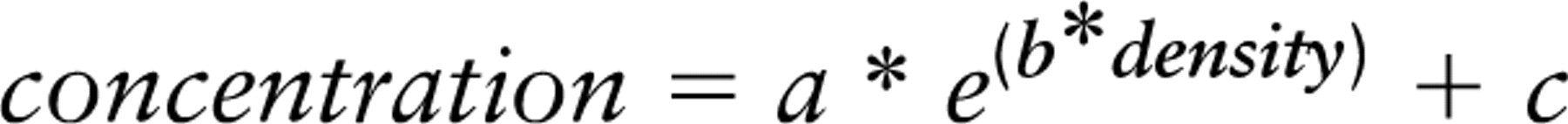
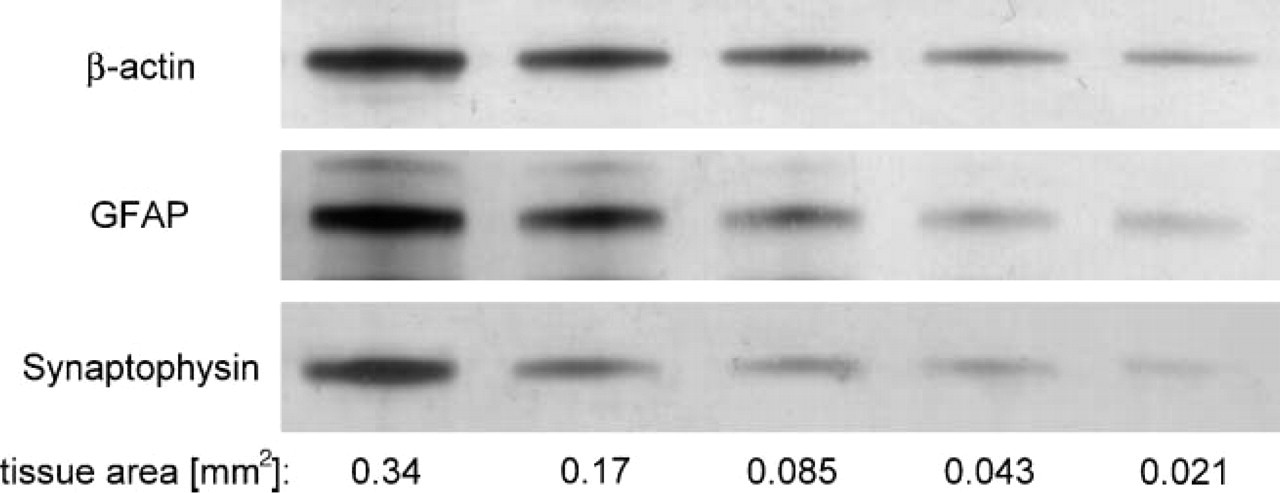
Western blot of a dilution series of a mouse brain section. An area of a mouse brain cryosection (20 μm) is diluted with section lysis buffer in logarithmic steps, subjected to SDS-PAGE, and blotted and immunodetected with an antibody against β-actin, glial fibrillary acidic protein (GFAP), and synaptophysin. The Western blot of the last five dilution steps is shown above with the corresponding size of area in the legend. An area of 0.021 mm2, corresponding to a tissue volume of 42 nl, is still detectable.
The area of the Nissl-stained section sufficient to conduct detection of β-actin is demonstrated in Figure 2. The area of 0.021 mm2 is roughly equivalent to 15 cortical neurons of mouse cortex.
Additionally, Western blots were performed from sections of paraffin-embedded tissue, which was fixed by fixation solution HOPE. Dewaxed paraffin sections revealed similarly strong bands of β-actin compared with the cryosections (data not shown).
Detecting Proteins in Different Brain Regions
Small areas of three different brain regions were investigated for expression of S-100 and GFAP in comparison to β-actin. In a section at a height of ∼1.5 mm caudal of bregma motor cortex, hippocampus and basal gang2lia were separated, each resulting in a size of ∼0.8 mm2. Western blots containing equal volumes of these samples are shown in Figure 3. S-100 protein revealed the strongest band in the tissue of basal ganglia (upper panel). Samples of both hippocampus and motor cortex showed a moderate intensity of S-100. Detection of GFAP of the mouse brain astrocytes resulted here in intense bands of basal ganglia and hippocampus (middle panel). A weak band is found in the sample of the cortical area. Intensities of the bands of β-actin are similar in all three areas (lower panel).
Digestion of Collected Tissue
The mouse prion protein has two moieties that are variably glycosylated. Figure 4 shows a Western blot with two lanes of tissue collected from the motor cortex at a height of bregma (each ∼1.5 mm2) that were incubated with the prion protein-specific antibody 6H4. A pattern of prion protein bands is visible in the left lane that includes the strongest band at ∼35 kDa and two bands with moderate intensity at ∼31 kDa and 27 kDa, which matches di-, mono-, and unglycosylated iso-forms. An additional band at 25 kDa is observed just below the unglycosylated band corresponding to a cleavage product of the prion protein with unknown glycosylation. In the right lane, the collected tissue was therefore digested prior to Western blotting with PNGase F, which cleaves all glycosylation moieties. A single strong band is found here at ∼27 kDa reflecting the (now unglycosylated) full-length prion protein, and a second band with moderate intensity is presented at ∼16 kDa, corresponding to the unmasked major cleavage product of the prion protein (formerly 25 kDa). Additional cleavage products cause bands with weak intensities that are just visible at ∼20 kDa. Thus, an additional application of digestion with PNGase to the sample allows deductions of the amounts and glycosylation of cleavage products.
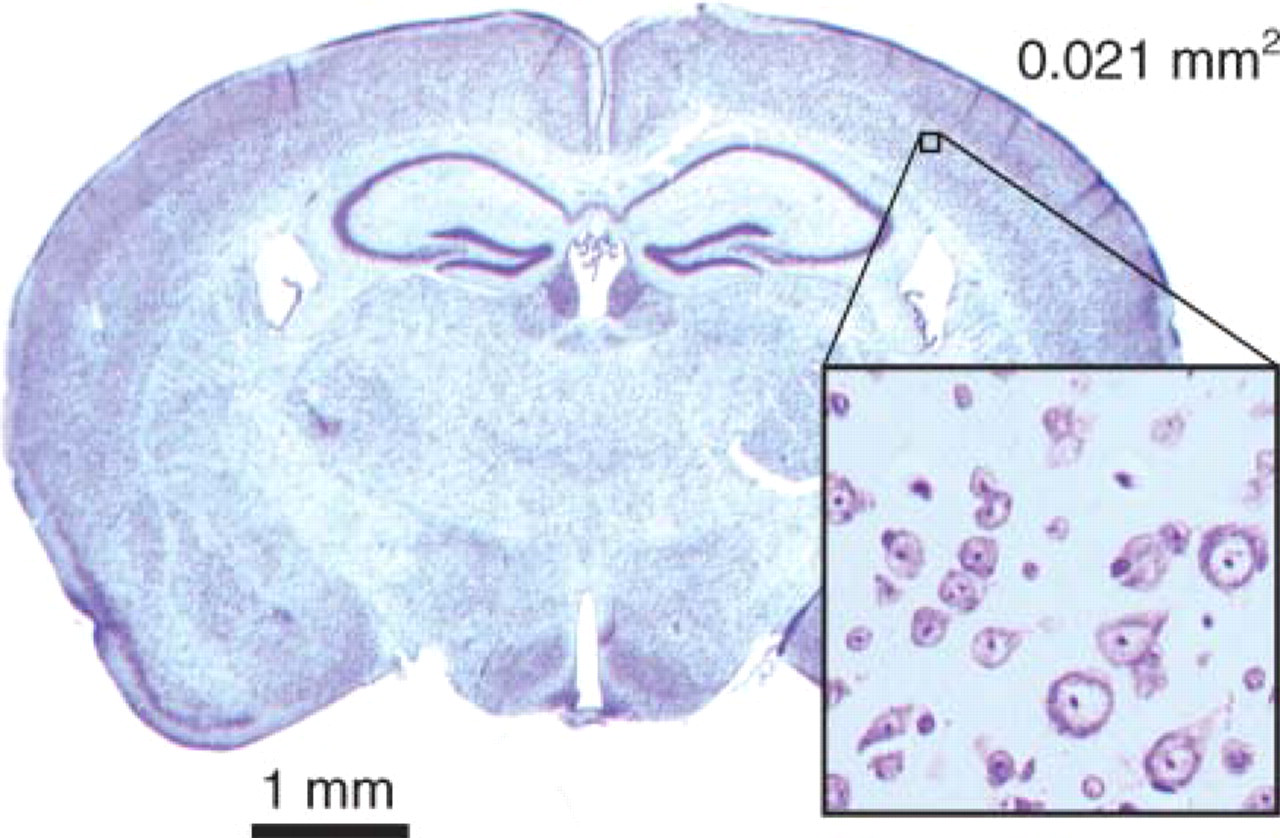
Nissl-stained section of a mouse brain. Enlarged section illustrates the minimal area detected in the Western blot dilution series (Figure 1) of a mouse brain section (0.021 mm2). The selected area of the cortex contains ∼15 cortical neurons.
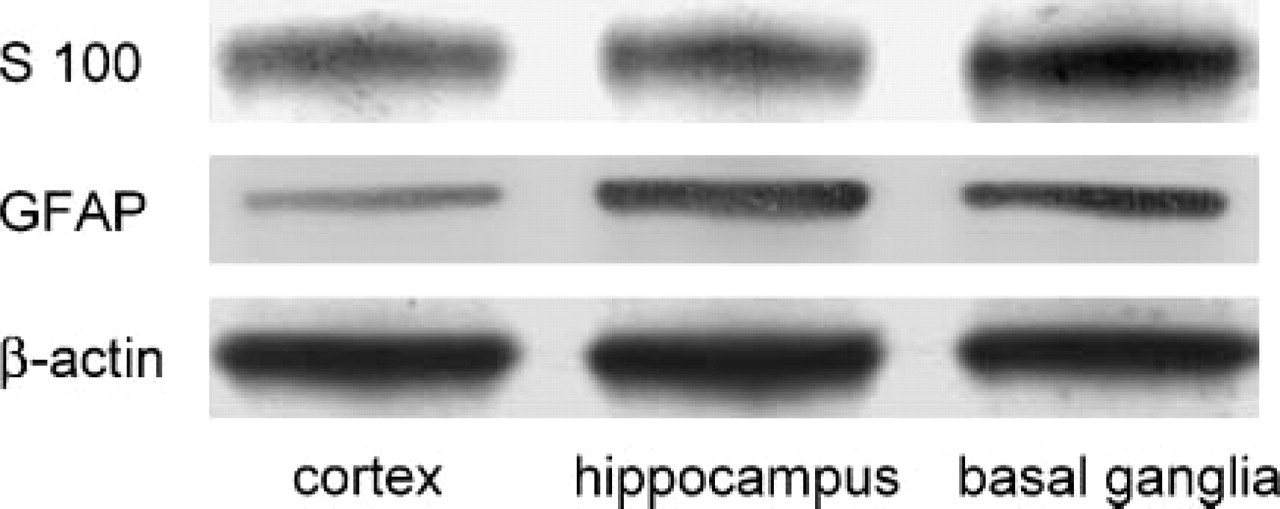
Different expression of proteins in histological regions of mouse brain. Western blots contain three samples each of cortex, hippocampus, and basal ganglia in equal volumes. Expression of S-100 is pronounced in basal ganglia, whereas cortex exhibits a low expression of the GFAP. β-actin is found to an equal extent in all three areas, representing the even load of tissue.
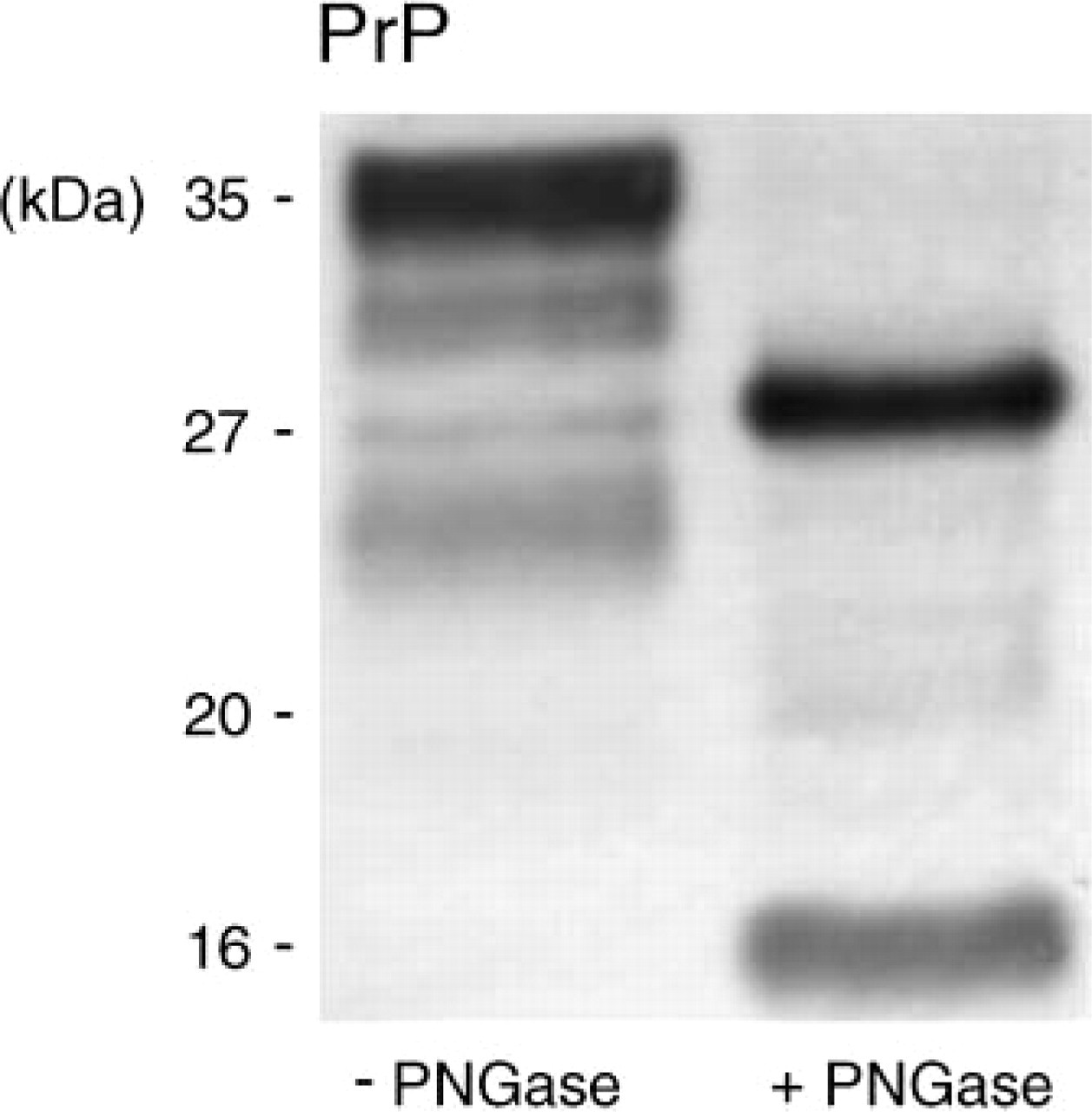
Normal cellular prion protein before and after deglycosylation. The prion protein contains two variable glycosylation moieties and undergoes a physiological cleavage. The left lane demonstrates the prion protein of an untreated sample from mouse cortex with three bands due to variable glycosylation and undefined cleavage products. The right lane contains an aliquot of the sample digested by PNGase. The full-length prion protein has a molecular weight of ∼27 kDa, whereas a second band presents a cleavage product of ∼16 kDa that was previously diglycosylated.
Western Blot of Phospho-Akt and Phospho-ERK
The activity of kinases is mostly represented by phosphorylation at distinct residues with an instable short-lived character. To test the conservation of quick phosphorylations, a neuronal cell line was left untreated or stimulated with insulin and overlaid with either conventional ice-cold Triton-based buffer or section lysis buffer. The conventional lysate had a cloudy aspect in contrast to the clear lysate with section lysis. After a short centrifugation step to remove any cell debris, Western blots were performed with equal volume loading. The phosphorylation of Akt at Ser473 and ERK 1,2 at Thr202/Tyr204 was detected in the samples with a phosphospecific antibody at 52 kDa and 42 kDa/44 kDa, respectively. Protein loading was controlled by a staining of β-actin. Samples based on section lysis buffer revealed the highest band intensities of phospho-Akt and phospho-ERK 1,2, but also β-actin, corresponding to a high amount of proteins. As shown in Figure 5, in the stimulated cells phosphorylation of Akt is clearly increased in comparison to the unstimulated cells presenting a low basal level (upper panel). The phosphorylation of ERK 1,2 is slightly raised in the stimulated cells, whereas the unstimulated cells have a well-detectable basal level (middle panel). Additionally, samples from (unstimulated) histological sections were tested, which reached low levels of phosphorylation in comparison to β-actin.
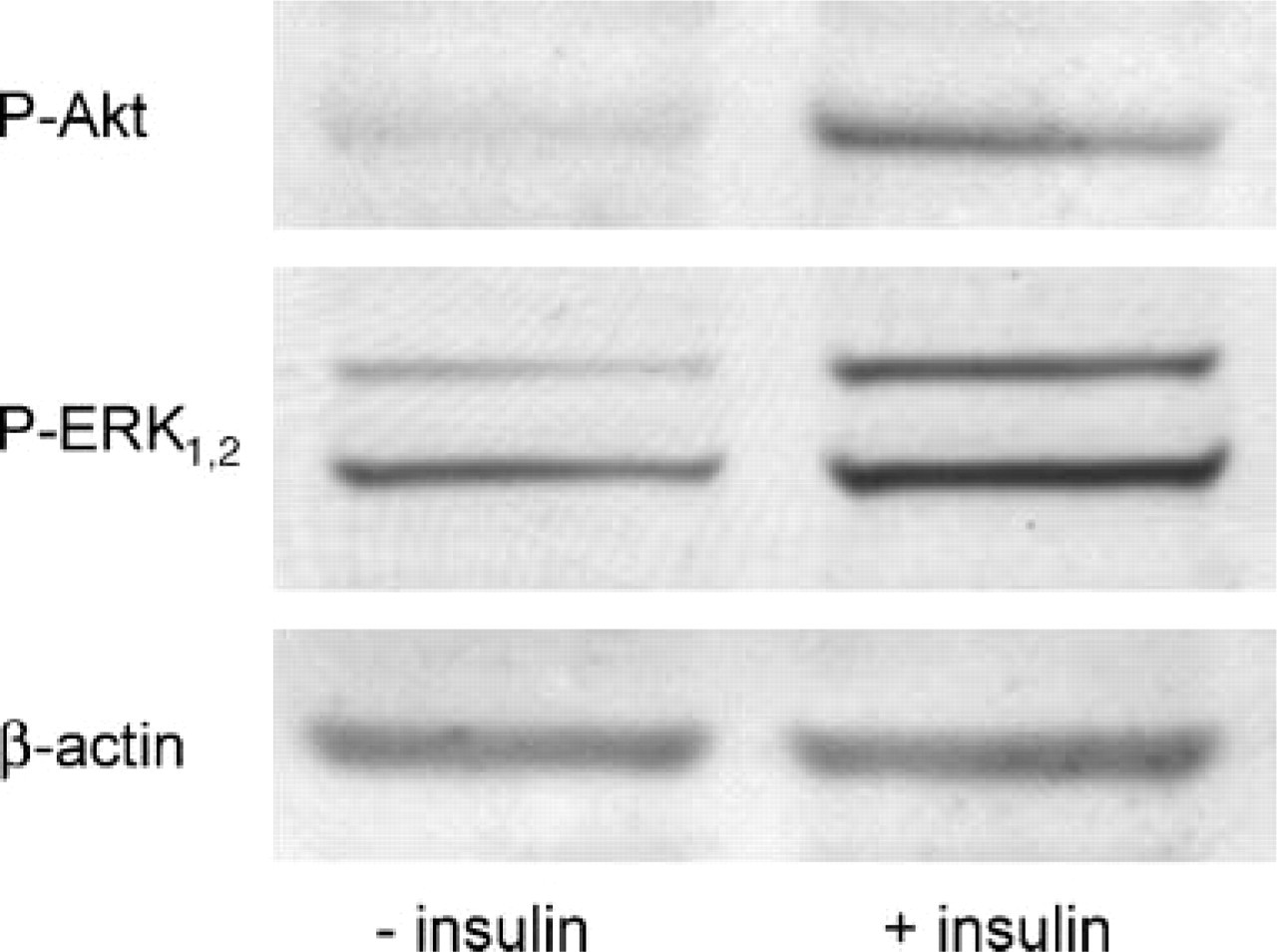
Detection of phosphorylation of Akt and ERK1,2 using section lysis buffer. Cell cultures of hippocampal neurons were left untreated or stimulated with insulin. Lysis was performed with section lysis buffer. Samples were subjected to Western blotting and detected with either phosphospecific Akt (upper panel) or phosphospecific ERK1,2 antibody (middle panel). In both samples clear phosphorylation of Akt and ERK1,2 was detected, whereby the stimulated cells (right lanes) revealed a stronger phosphorylation particularly regarding Akt. Equal protein loading was controlled by staining of β-actin (lower panel).
Discussion
The method of performing Western blots from histological sections provides an excellent tool for investigations of subregions of the brain. In the past, developments in Western blotting techniques mainly focused on increased sensitivity and emerged as an alternative to radioactivity. The indirect immunodetection by enzyme-coupled secondary antibodies allows generating enhanced fluorescing signals (chemoluminescence) (Whitehead et al. 1979; Thorpe et al. 1985; Bronstein et al. 1989) and an easy quantification by densitometry due to its linearity (Fradelizi et al. 1999). In this study, enzymatic staining within the membrane is used, representing a highly sensitive method as well. The relation between concentration of proteins and stain intensity can be expressed by a logarithmic function, thus spanning a wide range of values. Due to the absent diffusion on the X-ray film, the bands are noticeably sharper and allow miniaturizing the gels. For example, β-actin was shown here to be detectable in an area corresponding to ∼15 cortical neurons in a histological section. However, it should also be noted that the detection depends on the affinity of the antibody and expression of the respective protein.
Detectable minimum tissue allows investigating sub-regions in histological sections but requires special handling such as cooling to dewpoint to avoid air drying. The small amounts of sample volumes were, in fact, almost completely collected from histological sections but are not suitable for homogenization or centrifugation. Therefore, the section lysis buffer has to be capable of complete solubilization. The utilized SDS results in a lysate without visible fragments or clouding within minutes. After boiling the lysate, it has to be assumed that proteins are entirely solubilized. In combination with a strong reducing environment with agents such as dithiothreitol, section lysis buffer denatures several proteases and inhibits their activity, resulting in a very satisfactory extraction of protein both from cryo-sections and dewaxed sections (Wang et al. 1996). The amount of proteins in the lysate can be estimated in different ways; however, Bradford protein assay or bicinchoninic acid assay requires aliquots that might exceed the sample volumes (Sapan et al. 1999). Therefore, we calculate the loaded tissue by measurement of the collected area multiplied with the thickness of the section that can be easily controlled by confocal laser microscopy. Additionally, the use of a dilution series in the Western blot allows quantifying the protein expression and its relative expression to housekeeping proteins like β-actin as a ubiquitous component of cytoskeleton (Herman 1993). Furthermore, the amount of proteins is detectable by staining with Coomassie blue, Ponceau S, and other reagents (Bickar and Reid 1992; Dunn 1999).
The method is demonstrated with three separated areas of a mouse brain section (motor cortex, hippocampus formation, and basal ganglia). The similar expression of β-actin found in all samples represents an even protein loading, whereas the slightly divergent expression of S-100 and GFAP might correspond to the properties of neuroanatomical structures. S-100 protein is an acidic calcium-binding protein located predominantly in astroglial cells and less in oligodendrocytes (but also neurons) and might be released upon brain damage. Due to its association with these cells, the S-100 stain is most pronounced in the tissue of basal ganglia. Detection of GFAP in the mouse brain is accentuated in activated astrocytes, which are mostly present in basal ganglia and hippocampus (Schachner 1982; Muramatsu et al. 2003; Chang et al. 2005).
Some approaches might require particular pretreatments as shown here by deglycosylation of the cellular prion protein. In the untreated sample, multiple bands occur, and it is not possible to distinguish cleaved fragments by their molecular weight because of their unknown glycosylation as well as overlapping glycosylation patterns. However, from the digestion with PNGase F, it is concluded that there is a major cleavage product of the cellular prion protein that (a) accounts for about half of PrP full length, (b) is mostly diglycosylated, and (c) should therefore not be mistaken for the unglycosylated PrP full length in untreated sample, which migrates slightly higher as a discrete band.
In addition, other pretreatments might be applicable, such as mixing DNA cleaving enzymes to the section lysis buffer (benzonase 200 U/ml) to reduce the mucilaginous character of concentrated samples. The use of collagenase might be of interest for collagen-rich organs. For optimal enzymatic treatment, components in the section lysis buffer have to be reconsidered, especially concerning the pH or the strong reducing properties. On the other hand, the latter has an impact on the conservation of phosphorylation sites by inhibiting phosphatases. The section lysis buffer used here resulted in clear detection of phosphorylation of Akt and ERK 1,2 by specific antibody and therefore overcomes the problems of inhibitors and cooling with conventional lysis buffers. Moreover, lysis with section lysis buffer yields more proteins than with conventional lysis buffers. Recent studies preserved activities with a similar process of denaturation in cultured cell lines (Gil et al. 2003). The advantage of this application is the easy spreading over adherent cell cultures. It should be mentioned that the removed brain additionally requires immediate freezing, cutting, and lysing because there might be a loss of phosphorylation by time, air drying, and optional staining procedures.
We have developed a new technique whose major advantages are a satisfactory anatomical resolution and processing parallel to histological and immunhistological investigations. The ability of quantification in Western blot is superior to those of immunohistochmistry. Molecular weights can be determined, pretreatment like enzymatic digestion applied, and phosphorylations conserved. In conclusion, the described method of Western blotting of discrete microscopic regions in combination with other analytical methods will provide new insights into morphologic and functional relationships in the field of neuroscience and might also be used for other tissues.
Footnotes
Acknowledgments
We are grateful to Gertrud Kwiatkowski for excellent technical support and Dorothee Rieger and Nicole Weber for proofreading the manuscript.