
Abstract
We compared the reliability between apoptosis detection methods, namely, the terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) method and formamide-induced DNA denaturation assay using a monoclonal antibody (MAb) to single-stranded DNA (ssDNA) (formamide-MAb assay). Reaction targets in these methods are different: the TUNEL method recognizes free 3′-OH DNA ends, whereas the formamide-MAb assay detects ssDNA itself (25-30 bp). We found that the formamide-MAb assay immunohistochemically detected apoptotic cells, whereas the TUNEL method detected apoptotic cells as well as mitotic and necrotic cells. The TUNEL method recognized not only 3′-OH DNA ends cleaved by DNase during apoptosis but also constitutive physiological nicking that occurs in DNA duplication and histone posttranslational modifications during mitosis and random DNA breaks during necrotic execution. By electron microscopy, the mean labeling density (the number of 3′-OH DNA ends/nuclear area) obtained by the TUNEL method was determined to be consistently higher than that (the number of ssDNAs/nuclear area) obtained by the formamide-MAb assay. On the basis of these findings, we conclude that the formamide-MAb assay was more specific than the TUNEL method for the detection of apoptotic cells using electron microscopy; however, the labeling intensity of the formamide-MAb assay was slightly weaker than that of the TUNEL method.
A
Cell proliferation is commonly detected using various immunohistochemical methods such as bromodeoxyuri-dine (BrdU), proliferating cell nuclear antigen (PCNA), and Ki-67 immunostainings. For morphologic identification of apoptosis, transmission electron microscopy (TEM) is the method generally used because the concept of apoptosis proposed by Kerr et al. (1972) was based on several ultrastructural characteristics, particularly condensed chromatin. However, recent studies report that the classification of programmed cell death based on nuclear morphology does not take into account death signaling pathways triggered by caspases (Jäättelä 2004) or caspase-independent systems (Lockshin and Zakeri 2004). On the other hand, gel electrophoresis of DNA into a ladder has been accepted as a method of detecting apoptosis because DNA degradation into nucleosomal units is one of the hallmarks of apoptotic cell death (Nagata 2000). However, a contradiction between chromatin condensation and DNA fragmentation exists, namely, apoptosis without DNA fragmentation has been reported (Sakahira et al. 1999). The degradation of nuclear DNA into nucleosomal units is used as the basis for the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) method. The TUNEL method is generally applied more often for the detection of apoptosis than TEM because the TUNEL method does not require special equipment or a complex sample preparation as in TEM. Recent studies using the TUNEL method have cast doubt on its specificity of detecting apoptosis because the TUNEL method detects not only apoptotic cells but also necrotic and mitotic cells (Grasl-Kraupp et al. 1995; Sit et al. 1997; Wolvekamp et al. 1998; Otsuki 2000,2004). The TUNEL method by TEM (TUNEL/TEM) was developed for retrospective studies using archival epoxyresin-embedded materials and enables the morphological discrimination of apoptotic cells among many cell types including necrotic or mitotic cells based on ultrastructural characteristics (Inoki et al. 1997; Hayashi et al. 1998; Otsuki 2000,2004). To date, several studies of psoriasis have shown that almost all proliferative keratinocytes are TUNEL positive, although psoriasis is a hyperproliferative disease (Laporte et al. 2000). Our previous study of psoriasis by TUNEL/TEM (Kawashima et al. 2004) clearly demonstrated that immunogold particles indicating the presence of free 3′-OH DNA ends are localized on the nuclear euchromatin of cells in the S phase and, therefore, may actually label the site of physiological nicking yielded during DNA replication.
DNA of condensed chromatin, which is one of the apoptotic characteristics, is particularly sensitive to thermal denaturation occurring at a temperature lower than that for non-apoptotic cells (Allera et al. 1997). This critical nature of apoptotic DNA leads to a new method of apoptosis detection, i.e., formamide-induced DNA denaturation assay using a monoclonal antibody (MAb) against single-stranded DNA (formamide-MAb assay) (Frankfurt and Krishan 2001). We report here the results of a comparative study between the formamide-MAb assay and TUNEL method. The former method is considered to be a superior technique for apoptosis detection.
Materials and Methods
Cell Culture and Cell Preparation
Jurkat cells derived from human leukemic T-cells were cultured in the RPMI 1640 medium supplemented with 10% bovine serum. Induction of apoptosis was initiated by incubation for 8 hr in medium containing 0.5 μg/ml actinomycin-D (Tsuruga et al. 2003). Untreated Jurkat cultures served as controls. NP3 (mouse B lymphoma cell line) cells heated at 47C were used as necrotic cells (Hayashi et al. 1998). Apoptotic, necrotic, and control cells including mitotic cells were immediately rinsed with 0.01 M PBS, pH 7.4, fixed with 1% paraformaldehyde in PBS for 10 min for light microscopy, mounted on aminopropyltriethoxysilane-coated glass slides, and subjected to the following treatments. For TEM, cells were fixed with 2.5% glutaraldehyde and 2% formaldehyde in 0.1 M phosphate buffer (pH 7.4) for 1 hr at 4C and then rinsed overnight with PBS at 4C. Cells were subsequently postfixed in 1% OsO4 for 45 min at room temperature (RT), dehydrated in a series of graded ethanol concentrations, cleared in propylene oxide, and embedded in an epoxy resin mixture (LUVEAK 812, LUVEAK MNA, LUVEAK DDSA, LUVEAK DMP-30; Nacalai Tesque Inc., Kyoto, Japan) under the conditions described by Luft (1961). Semithin sections stained with toluidine blue were used to confirm the presence of apoptosis or necrosis in treated cells. Ultrathin sections (70-80 nm) were prepared and mounted on nickel grids (Stork Veco International Inc.; Bedford, MA).
Evaluation of TUNEL Method with Light Microscopy (TUNEL/LM)
The TUNEL method was conducted according to the manufacturer's protocol (Apop Tag Apoptosis Detection Kit; Chemicon International, Temecula, CA). Briefly, cells were postfixed in the 2:1 (v/v) mixture of ethanol and acetic acid at −20C, endogenous peroxidase was quenched with 3% H2O2 in PBS for 5 min, and cells were then incubated with deoxynucleotides conjugated to digoxigenin, which was bound to 3′-OH DNA ends by exposing cells to the TdT enzyme for 1 hr at 37C. After 3′-OH DNA end labeling, cells were further incubated with an anti-digoxigenin antibody conjugated to horseradish peroxidase for 1 hr at RT. Cells were reacted with 0.05% 3-3′-diaminobenzidine tetrahydrochloride (DAB) for 5 min and then counterstained with 1% methyl green.
Evaluation of Formamide-MAb Assay with LM (Formamide-MAb/LM)
Cells were incubated in saponin (0.1 mg/ml in PBS; Nacalai Tesque Inc.) and treated with proteinase K (20 μg/ml in PBS; Roche Diagnostics GmbH, Mannheim, Germany) for 20 min at RT. The cells were incubated in 50% (v/v) formamide in distilled water at 56C for 20 min and then transferred to cold PBS for 15 min, followed by exposure to Block Ace (Dainippon Sumitomo Pharma Co., Ltd.; Tokyo, Japan) for 20 min to block nonspecific antibody binding. Cells were then incubated overnight with a primary mouse anti-ssDNA MAb (clone F7-26, cat. #MAB3299; Chemicon International) at a dilution of 1:50. Cells were incubated with a secondary biotinylated anti-mouse immunoglobulin for 60 min and then incubated with streptavidin conjugated to horseradish peroxidase for 30 min. After the application of DAB solution, cells were then counterstained with 1% methyl green.
TUNEL- or formamide-MAb-positive cells were quantified as the ratio of the number of TUNEL- or formamide-MAb-positive cells to the total number of cells, which was obtained in >10 optical fields at × 200 magnification. Differences in the numbers of TUNEL- or formamide-MAb-positive control, apoptotic, and necrotic cells were analyzed using Student's t-test.
Evaluation of TUNEL/TEM
TUNEL/TEM has been described in detail elsewhere (Inoki et al. 1997; Otsuki and Ito 2001). An etching step using saturated sodium metaperiodate was performed for 2 min, followed by rinsing in distilled water for 15 min. The TUNEL method was conducted according to the manufacturer's protocol (Apop Tag Apoptosis Detection kit; Chemicon International); briefly, deoxynucleotides conjugated to digoxigenin were bound to the 3′-OH DNA ends by exposing grid mounted sections to the TdT enzyme for 1 hr at 37C. After 3′-OH DNA end labeling, sections were further incubated with an anti-digoxigenin antibody conjugated to 10-nm colloidal gold particles (Aurion Co.; Wageningen, The Netherlands) for 1 hr at RT. After staining with uranyl acetate and lead citrate, sections were examined by TEM using H-7100 (Hitachi; Tokyo, Japan).
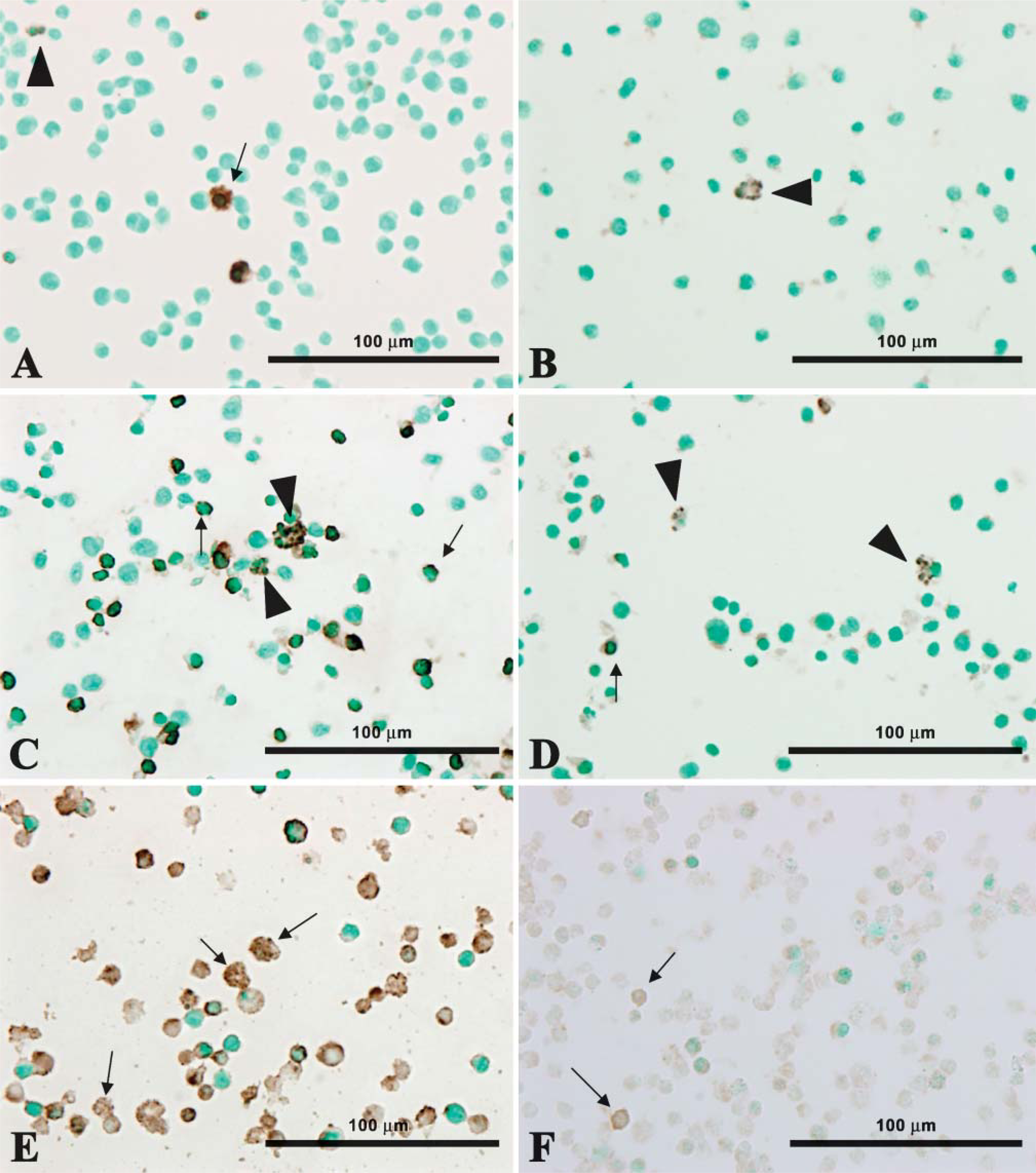
TUNEL/LM and formamide-MAb/LM. The quiescent (arrow) and non-quiescent (arrowheads) nuclei of control cells are TUNEL/LM (
Evaluation of Formamide-MAb Assay with TEM (Formamide-MAb/TEM)
Etching was performed as described for TUNEL/TEM. After rinsing once in distilled water, the grid-mounted ultrathin sections were incubated in 50% (v/v) formamide in distilled water at 56C for 5 min. Sections were then transferred to cold PBS for 15 min, followed by exposure to 20% normal goat serum for 20 min to block nonspecific antibody binding. Sections were then incubated with a mouse anti-ssDNA MAb at a dilution of 1:50 with PBS containing 10% normal goat serum for 1 hr. Sections were incubated with a mouse IgM secondary antibody conjugated to 10-nm immunogold particles (British BioCell International; Golden Gate, UK) for 30 min. Sections were stained again with uranyl acetate and lead citrate and examined by TEM (Hitachi).
Image Analysis for Free 3′-OH DNA Ends and ssDNA
Image analyses of free 3′-OH DNA ends and ssDNA were performed using the NIH Image program (http://rsb.info.nih.gov/nih-image/download.html). Electron microscopy images of Jurkat or NP3 cell nuclei that were subjected to TUNEL/TEM or formamide-MAb/TEM were scanned into the computer. We then determined labeling density using both the NIH Image program for nuclear area (μm2) and the number of immunogold particles in the nuclei of more than 10 Jurkat cells or 10 NP3 cells of each sample. Differences in the labeling density of 3′-OH DNA ends or ssDNA among the control, mitotic, and apoptotic cells were analyzed statistically using Student's t-test or Welch's t-test.
Results
Evaluation of TUNEL/LM and Formamide-MAb/LM
There were only a few TUNEL/LM- and formamide-MAb/LM-positive cells among control cells (Figures 1A and 1B). In apoptotic Jurkat cells, round, crescent, and fragmented nuclei were contained in TUNEL/LM-or formamide-MAb/LM-positive cells. (Figures 1C and 1D). The number of TUNEL/LM-positive cells was much larger than that of formamide-MAb/LM-positive cells. Heat treatment made the cytoplasm of NP3 cells swollen and the nuclei of NP3 cells transparent (Figures 1E and 1F), and the nonspecific staining of the cytoplasm caused by nuclear membrane rupture was more intense in TUNEL/LM than in formamide-MAb/LM.
Quantitative Analysis by TUNEL/LM and Formamide-MAb/LM
Ratios of TUNEL/LM- and formamide-MAb/LM- positive cells in control, actinomycin-D-treated, and heated NP3 cells are shown in Figure 2. Ratio of TUNEL-positive cells was always significantly higher than that of formamide-MAb-positive cells in all the normal, apoptosis, and necrosis models (Figure 2).
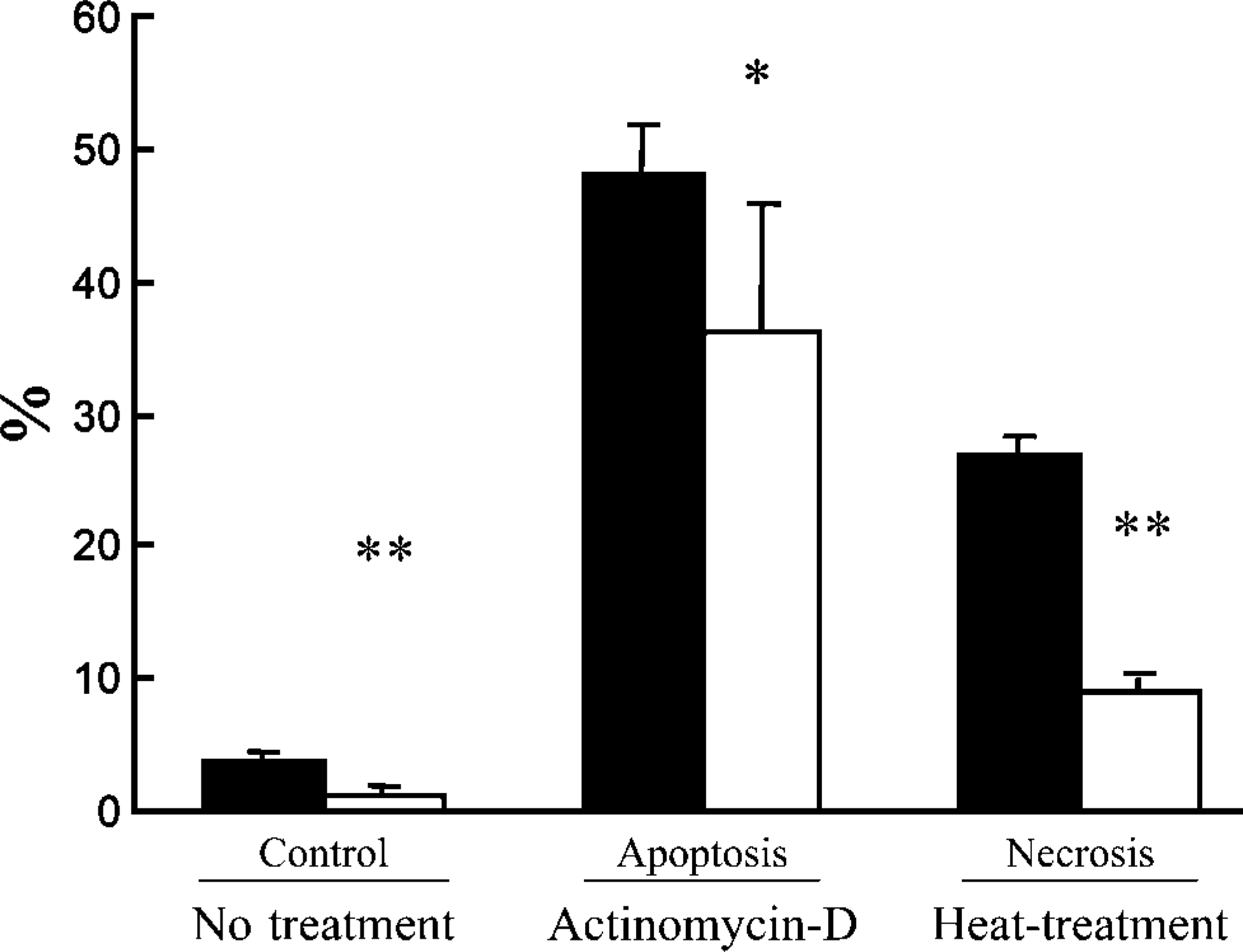
Quantitative analyses by TUNEL/LM (black columns) and formamide-MAb (white columns) using control, apoptotic Jurkat cells treated with actinomycin-D and necrotic NP3 cells heated at 47C. Data presented are means ± SD. ∗ p<0.01 and ∗∗ p<0.001 indicate significant differences between ratios of TUNEL/LM- and formamide-MAb-positive cells.
Cell Morphology
Control cells had euchromatin-rich pale nuclei with prominent nucleoli, sometimes showing mitotic nuclei (Figure 3A). On the other hand, the majority of Jurkat cells treated with actinomycin-D (0.5 μg/ml) were characterized by crescent or rounded nuclei consisting of condensed chromatin that showed a strong intensity of staining with toluidine blue (Figure 3B). Almost all the heat-treated NP3 cells showed swelling, ruptured cell membrane, and vacuoles in the cytoplasm and an increased intensity of staining of heterochromatin in the nuclei (Figure 3C).
TUNEL/TEM
TUNEL/TEM revealed the distribution of some immunogold particles on the even nuclear euchromatin of control cells (Figure 4A). There was also an intense staining of the condensed chromatin in cells undergoing mitosis (Figure 4B). In apoptotic cells induced by actinomycin-D, the rounded, condensed nuclear chromatin contained many immunogold particles indicating free 3′-OH DNA ends (Figure 5A). Necrotic cells induced by heating at 47C are characterized by ruptured membranes, degenerative organelles, and the condensation of chromatin in the nuclei, as well as apoptotic cell nuclei, and contained many immunogold particles (Figure 5B).
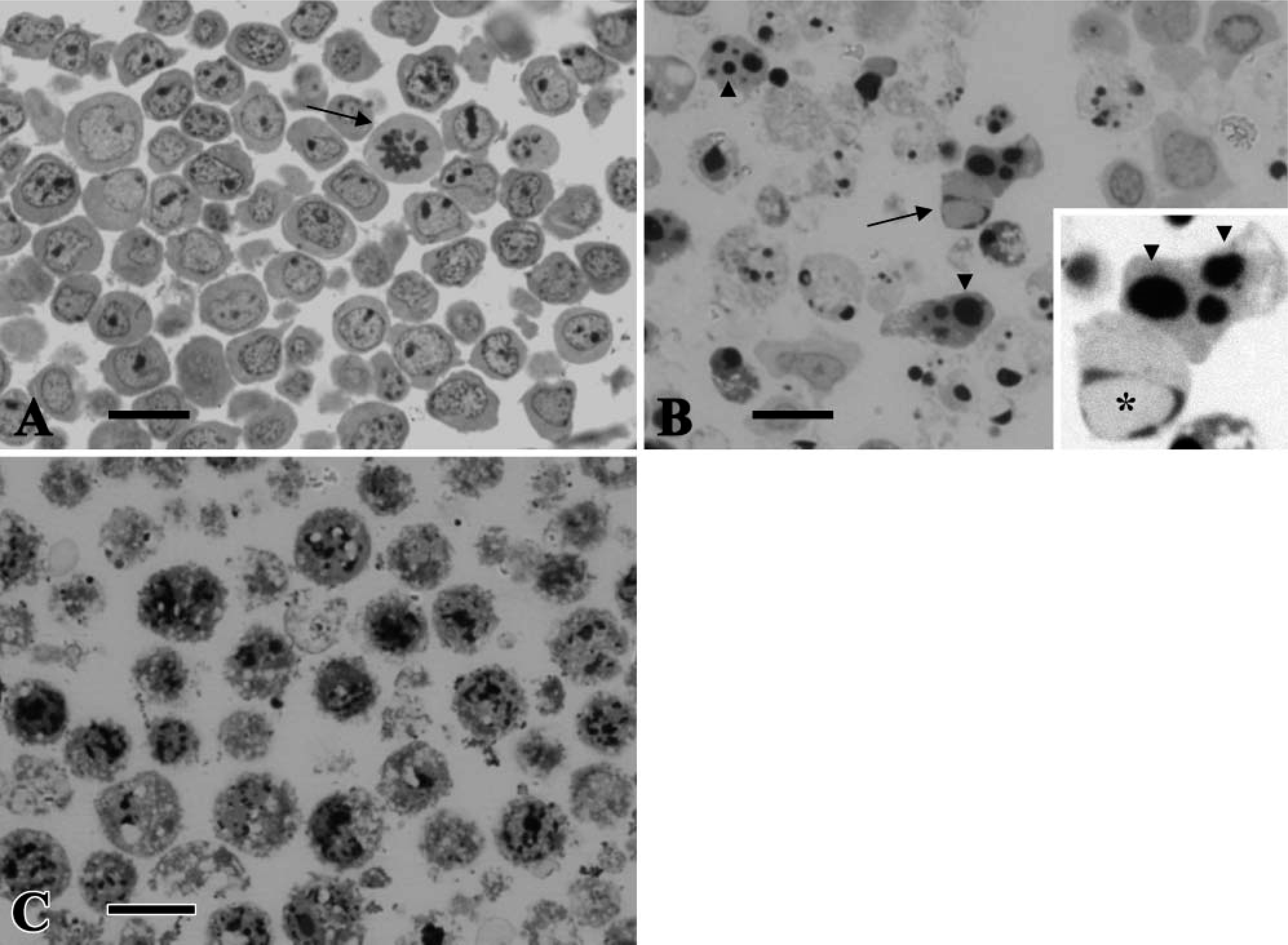
Semithin sections of epon-embedded samples stained with toluidine blue. Control Jurkat cells (
Formamide-MAb/TEM
In contrast to TUNEL/TEM, very few immunogold particles indicating the existence of ssDNA were observed in either quiescent or mitotic nuclei of control cells (Figures 4C and 4D). In apoptotic cell nuclei, however, immunogold particles were evenly distributed on the condensed chromatin of the nuclei, suggesting a greater ssDNA formation associated with apoptosis (Figure 5C). In the nuclei of necrotic cells, only a few immunogold particles were distributed on the agglutinated heterochromatin (Figure 5D).
Quantitative Analyses by TUNEL/TEM and Formamide-MAb/TEM
Statistical analysis of the labeling density obtained by TUNEL/TEM and formamide-MAb/TEM in the nuclei of control, mitotic, apoptotic, and necrotic cells is shown in Table 1. Mean labeling densities obtained by TUNEL/TEM in control, mitotic, apoptotic, and necrotic cells were 0.7 ± 0.2/μm2, 10.9 ± 0.1/μm2, 19.5 ± 5.0/μm2, and 6.9 ± 0.5/μm2, respectively. Mean labeling densities obtained by TUNEL/TEM were significantly higher in the mitotic, apoptotic, and necrotic cells than in control cells (p<0.001). Mean labeling densities obtained by formamide-MAb/TEM in the control, mitotic, apoptotic, and necrotic cells were 0.9 ± 0.4/μm2, 1.5 ± 0.5/μm2, 23.1 ± 8.3/μm2, and 1.3 ± 0.6/μm2, respectively. Mean labeling density obtained by formamide-MAb/TEM was consistently higher in apoptotic cells than in control, mitotic, and necrotic cells, whereas no statistically significant difference was found among control, mitotic, and necrotic cells. Mean labeling densities of mitosis and necrosis obtained by formamide-MAb/TEM were lower than that obtained by TUNEL/TEM. However, labeling densities of apoptotic cells obtained by formamide-MAb/TEM were higher than that obtained by TUNEL/TEM, and the difference in mean average count between the methods was not statistically significant (Table 1).
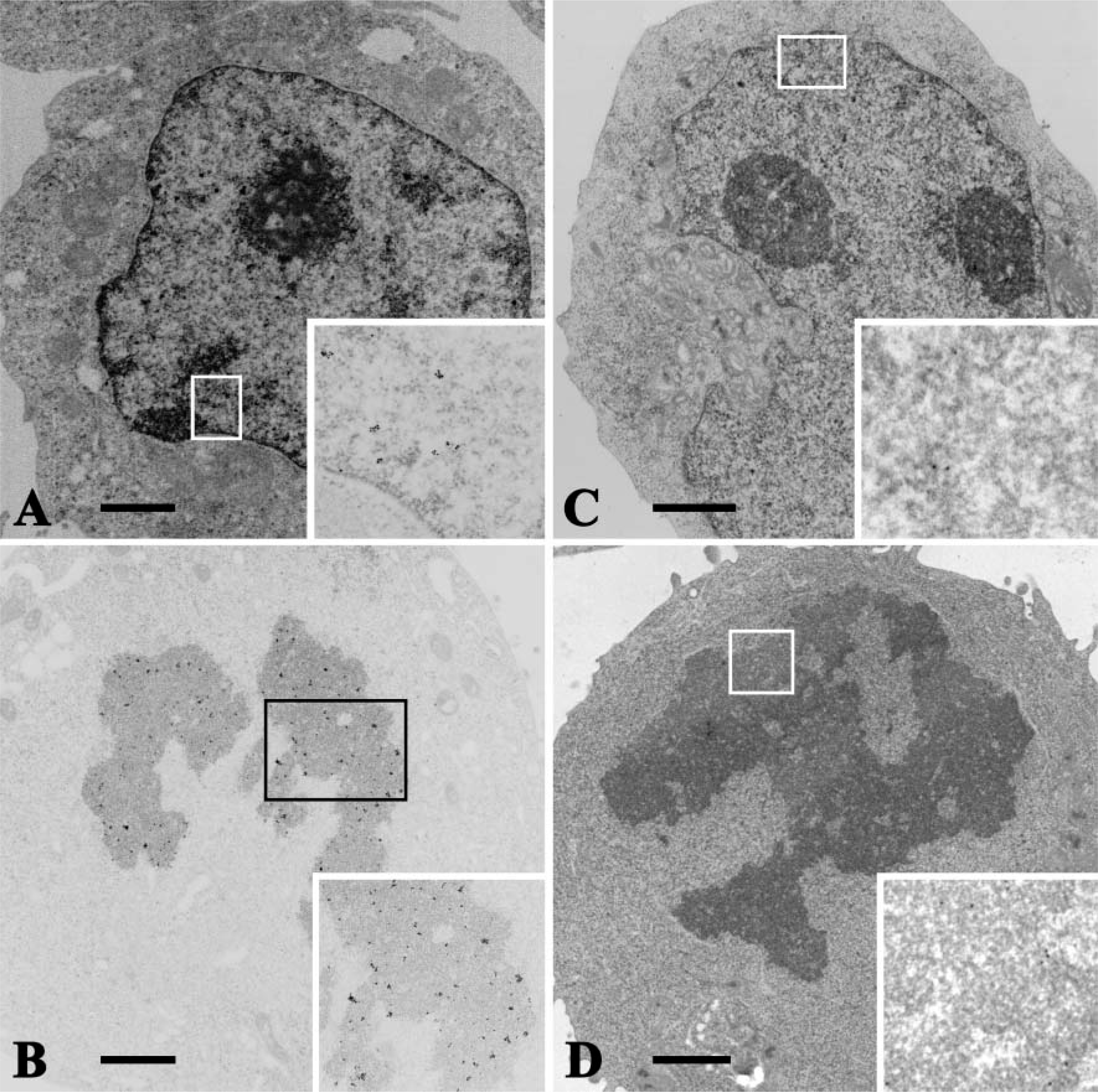
TUNEL/TEM and formamide-MAb/TEM. Normal quiescent (
Discussion
In this study we use actinomycin-D to induce apoptosis in Jurkat cells. Actinomycin-D is widely used as an apoptosis inducer via transcription inhibition, following DNA damage (Tsuruga et al. 2003). This DNA damage induces caspase-dependent compaction and fragmentation of chromatin into crescent-shaped or spherical masses at the nuclear periphery. Recent apoptosis signaling pathway studies have shown caspase-independent apoptosis (Lockshin and Zakeri 2004) and three subtypes of apoptosis according to nuclear morphology (Jäättelä 2004). The first type is classic apoptosis characterized by condensation of chromatin into compact masses. The second type is apoptosis-like programmed cell death (PCD or active cell death), for example, caspase-independent chromatin margination triggered by apoptosis-inducing factor. Furthermore, the serine protease activity of Omi/Htra2 mediates caspase-independent cellular rounding and shrinkage without changes in nuclear morphology. The third type is necrosis-like PCD in which Ca2+ and reactive oxygen species lead to mitochondrial dysfunction inducing PCD. Further studies are needed to clarify the relationship between nuclear morphology and apoptosis signaling pathways. So far, many apoptosis detection methods have been developed: JC-1 staining (permeabilization of the mitochondrial membrane or mitochondrial membrane potential), annexin V staining (appearance of phosphatidylserine in the outer leaflet of the plasma membrane), and fluorescense dyes, flow cytometry, TUNEL method, and DNA agarose gel electrophoresis (condensation and fragmentation of the chromatin). In addition, a modified TUNEL method recently developed using BrdU with propidium iodide for flow cytometry may give specificity for apoptosis detection. But flow cytometry cannot show morphology of apoptotic cells. Researchers will be able to choose optimum methods in their experiments, and several methods have been proposed for determination of apoptosis.
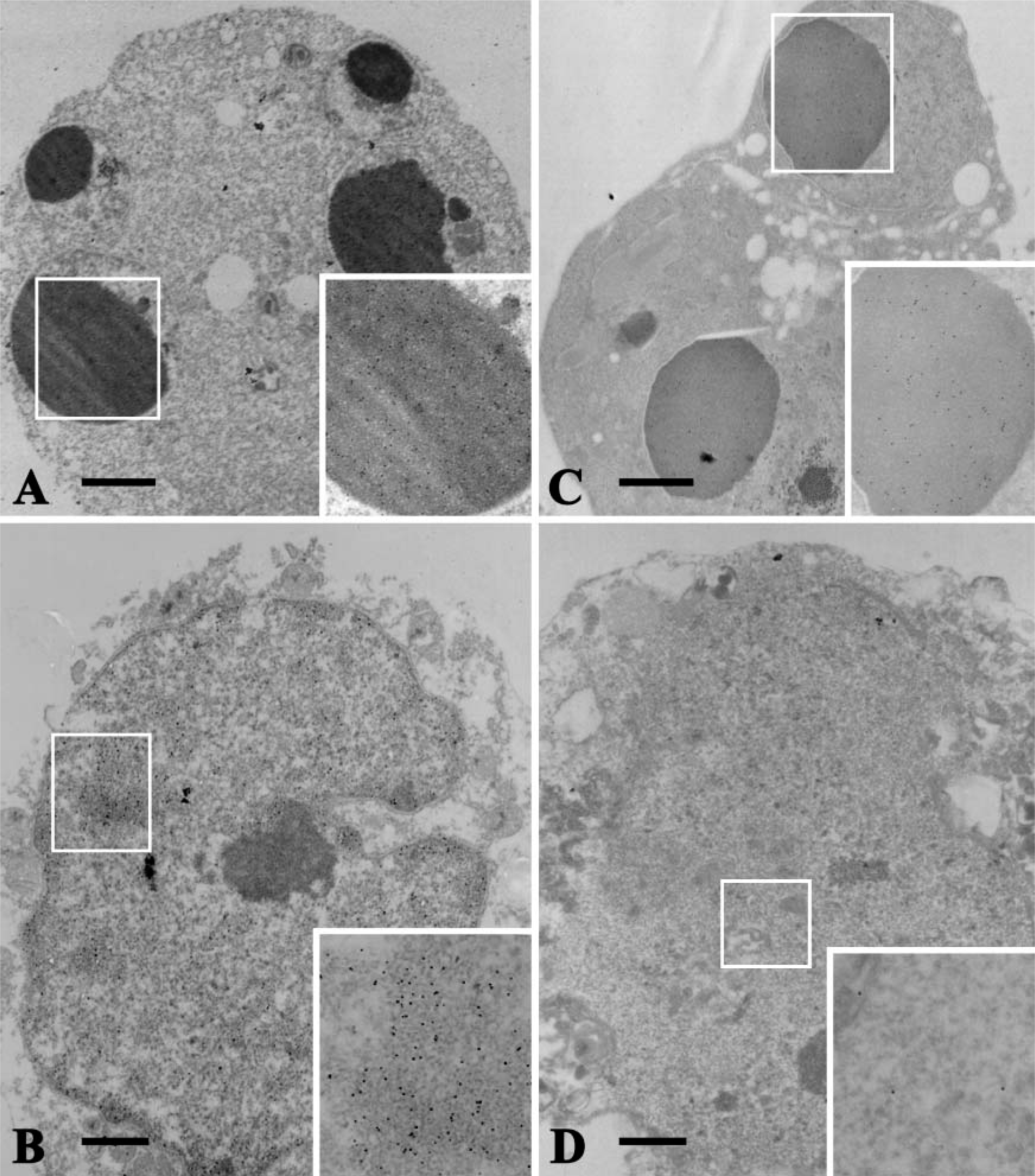
TUNEL/TEM and formamide-MAb/TEM. Alarge number of immunogold particles were observed on the condensed chromatin of an apoptotic cell (
A question concerning the specificity of the TUNEL method for detecting apoptosis has been raised because it detects not only apoptotic cells but also necrotic and mitotic cells (Grasl-Kraupp et al. 1995; Sit et al. 1997; Wolvekamp et al. 1998; Otsuki 2000,2004), and it recognizes only free 3′-OH DNA ends. In necrotic cells, however, free 3′-OH DNA ends are also generated. Nevertheless, TUNEL/TEM has many advantages over TUNEL/LM. First, the former enables retrospective examination of Epon-embedded specimens previously prepared for routine TEM. Second, it enables the identification of preapoptotic cells based on their ultra-structural appearance (Migheli et al. 1995; Negoescu et al. 1996; Sanders and Wride 1996; Inoki et al. 1997; Hayashi et al. 1998; Ohno et al. 1998; Gertrud et al. 1999; Saitoh et al. 2000; Barth et al. 2002). Third, it enables the quantitative analysis of free 3′-OH DNA ends in special cases such as in epidermal keratinization, in which apoptosis occurs without the formation of apoptotic bodies (Hayashi et al. 1998; Nakamura et al. 1999; Okamura et al. 2002). Fourth, with some modifications, it shows DNA fragmentation occurring at 5′-OH and 3′-OH termini in apoptotic cells (Hayashi et al. 1998; Otsuki and Ito 2001). Using TUNEL/TEM, some researchers found immunogold particles, indicative of nicked 3′-OH ends, distributed on the euchromatin of proliferating cells (Migheli et al. 1995; Sanders and Wride 1996; Inoki et al. 1997; Hayashi et al. 1998; Nakamura et al. 1999). We also observed immunogold particles on the nuclear euchromatin of control cells in the present study. There are some possible reasons for the labeling of normal euchromatin, such as the presence of artifacts generated during etching, which involves oxidation (Ansari et al. 1993), the physical exposure of 3′-OH ends due to ultrathin sectioning (Ansari et al. 1993), and the presence of DNA breaks induced by DNA repair systems or sites of active gene transcription located in the chromatin (Thiry 1991). In this study, next to 3′-OH DNA ends having a high labeling density was the mitotic nuclei. DNA duplication was completed in mitotic chromosomes, and the condensation of chromatin was enhanced by the condensin protein that binds to DNA. Histone posttranslational modifications such as phosphorylation occur on the linker protein histone 1 (H1) and on the core protein histone 3 (H3). Phosphorylation of H1 may weaken the association of H1 with chromatin and enable other condensation-inducing factors to interact with the chromatin (Hirano 2000). The treatment of sections, such as etching with saturated sodium metaperiodate, might nick DNA at the linker protein H1, resulting in free 3′-OH DNA ends that were detected by TUNEL/TEM.
Quantitative analysis of labeling density by TUNEL/TEM and formamide-MAb/TEM

a,b p<0.001. The mean labeling densities in mitotic and necrotic cells determined by formamide-MAb/TEM were lower than those determined by TUNEL/TEM.
p<0.001. The mean labeling density of formamide-MAb/TEM was consistently higher in the apoptotic cells than in the control, mitotic, and necrotic cells. These cells were treated as described in the legend of Figure 2. Data presented are means ± SD.
Frankfurt et al. (1996) developed a new immunohistochemical method for light microscopy detection of apoptosis using a MAb to ssDNA. This technique specifically enables the identification of apoptotic cells—not chemically induced necrotic cells—even in the early stages of apoptosis. Other groups confirmed that, with the use of their prepared antibodies, ssDNA immunohistochemistry is more effective than TUNEL for the detection of apoptosis (Naruse et al. 1994; Watanabe et al. 1999). They further demonstrated that the number of ssDNA-positive cells is far smaller than that of TUNEL-positive cells in most samples examined by both methods. Recently, Frankfurt and Krishan (2001), using formamide in conjunction with a MAb to ssDNA, have further improved the methodology on the basis of the biological nature of apoptotic DNA, i.e., the low denaturing temperature of intact apoptotic DNA. At low temperatures, formamide induces DNA stretching and breaking, allowing a MAb to specifically bind to deoxycytidine in the 25- to 30-base ssDNA. In the present study, although the mean ssDNA labeling density in apoptotic nuclei was significantly higher than in the control and mitotic nuclei, the mean ssDNA labeling density in the mitotic nuclei was not significantly different from the control cells. Whereas the phosphorylation of H3 results in a tight chromatin condensation in mitotic nuclei, the deacetylation of H3 and H4 during apoptotic chromatin condensation decreases the affinity of histones for DNA (Allera et al. 1997); therefore, formamide treatment readily provides ssDNAs.
In our previous study using TUNEL/TEM (Hayashi et al. 1998), although early-stage necrotic cells demonstrated a higher 3′-OH/5′P labeling density, this density decreased at the late stage. Although some immunogold particles in necrotic cell nuclei may indicate DNA cleavage sites at 3′-OH DNA ends, various endonucleases may be activated during these necrotic processes, in addition to lysosomal DNases that randomly degrade DNA, resulting in DNA strand breaks (Harmon et al. 1990). The quantitative analyses in the present study showed that the density of labeling of 3′-OH ends was higher than that of ssDNA not only in necrotic nuclei but also in control, mitotic, and apoptotic nuclei, which agrees with the data of Frankfurt and Krishan (2001). Formamide-MAb assay, i.e., DNA denaturation assay, is based on changes in chromatin associated with condensation induced by formamide and is not influenced by DNA strand breaks. Groos et al. (2003) evaluated seven different techniques for the in situ detection of apoptosis and also concluded that TUNEL is less reliable than the new formamide-MAb assay. Proliferating cells in psoriatic skin are evidently TUNEL positive (Kawashima et al. 2004), but these cells are not considered to be apoptotic as assessed by the formamide-MAb assay, in agreement with the study of Takahashi et al. (2002).
In formamide-MAb/TEM, apoptotic or necrotic cells were identified electron microscopically, and the number of immunogold particles was counted (Table 1). The number of immunogold particles in necrotic cells determined by TUNEL/TEM was much higher than that determined by formamide-MAb/TEM, in agreement with previous reports (Frankfurt et al. 1996; Frankfurt and Krishan 2001).
In conclusion, in the case of TEM of ultrathin sections, the formamide-MAb assay appears to offer a higher degree of specificity for detecting apoptotic cells than the TUNEL method. Moreover, it was able to avoid false positivity to apoptotic cells in proliferating cells of the tissues.
Footnotes
Acknowledgements
This work was supported in part by a Grant-in-Aid for General Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 14571592).