
Abstract
In this article we describe a novel effect of formamide on DNA of apoptotic nuclei and present a method for specific detection of apoptotic cells based on this effect. Our observations show that formamide induces DNA denaturation in apoptotic nuclei but has no such effect on DNA of non-apoptotic cells. Formamide-induced DNA denaturation combined with detection of denatured DNA with a monoclonal antibody (MAb) against single-stranded DNA made it possible to specifically identify the apoptotic cells. This procedure produced intense staining of the condensed chromatin in the apoptotic nuclei. In contrast, necrotic cells from cultures treated with sodium azide, saponin, or hyperthermia did not bind this antibody, demonstrating the specificity of the formamide–MAb assay for the apoptotic cells. However, TUNEL stained 90–100% of necrotic cells in all three models of necrosis. Because the MAb did not stain cells with single- or double-stranded DNA breaks in the absence of apoptosis, we conclude that staining of the apoptotic nuclei is not influenced by DNA breaks and is induced by specific changes in condensed chromatin, such as damage to the DNA–histone interactions. Importantly, the formamide–MAb technique identified apoptotic cells in frozen sections and in histological sections of formalin-fixed, paraffin-embedded tissues.
Keywords
A
Biochemical and cytochemical studies have demonstrated that DNA of apoptotic chromatin has increased sensitivity to thermal denaturation. Analysis of nuclei by scanning calorimetry to detect DNA denaturation and by electrophoresis to evaluate DNA molecular weight demonstrated that intact DNA of apoptotic cells denatured at a lower temperature than that of non-apoptotic cells. (Allera et al. 1997). These data indicated that increased sensitivity to thermal denaturation in apoptotic DNA is not induced by DNA breaks and may reflect specific changes in the apoptotic chromatin.
Monoclonal antibodies (MAbs) to single-stranded DNA (ssDNA) were used in our previous studies to evaluate DNA denaturation in situ. These MAbs require a large stretch of ssDNA for the binding and do not interact with DNA breaks without heating (Frankfurt 1990; Frankfurt et al. 1996). Heating induced DNA denaturation in cells with drug-induced DNA breaks and in apoptotic cells. Denatured DNA was detected with an MAb specific to ssDNA. Intensity of MAb binding was significantly higher in the apoptotic cells, and under certain conditions selective denaturation of apoptotic DNA was induced (Frankfurt 1994; Frankfurt et al. 1996).
The goal of the present study was to identify conditions that induce DNA denaturation in apoptotic cells but not in necrotic cells or in cells with DNA breaks in the absence of apoptosis. By using a gentle DNA denaturing agent, formamide, and heating at low temperature, we were able to induce DNA denaturation only in condensed apoptotic chromatin. To determine whether the binding of an anti-ssDNA MAb under these conditions is indeed specific to the apoptotic cells, the following models were tested: (a) apoptosis not accompanied by internucleosomal DNA fragmentation; (b) necrosis with a high level of double-stranded DNA breaks detected by TUNEL; (c) cells with single-stranded DNA breaks induced by oxygen radicals; and (d) cells with double-stranded DNA breaks induced by DNase.
From these studies, we conclude that formamide-induced DNA denaturation in the apoptotic cells is not influenced by DNA breaks but reflects changes in DNA stability induced by specific changes in condensed chromatin, such as damaged DNA–histone interactions. These data demonstrated that formamide-induced DNA denaturation and staining with an anti-ssDNA MAb could detect condensed apoptotic chromatin and can be used for identification of apoptotic cells. Importantly, the formamide–MAb procedure specifically stained the apoptotic cells and clearly distinguished apoptosis from necrosis. This method identified apoptotic cells in frozen sections and in sections of formalin-fixed, paraffin-embedded tissues. Preliminary data were published in abstract form (Frankfurt and Krishan 2000).
Materials and Methods
Reagents
Culture medium, heat-inactivated fetal bovine serum (FBS), trypsin–EDTA, S1 nuclease, and proteinase K were purchased from Gibco (Grand Island, NY). Fluorescein-conjugated goat antimouse IgM was obtained from Sigma (St Louis, MO) and peroxidase-conjugated rat monoclonal anti-mouse IgM from Zymed (San Francisco, CA). Apostain (Miami, FL) supplied MAb F7–26 specific to ssDNA. Staurosporine, formamide, propidium iodide, and 4–6-diamidino-2-phenylindole (DAPI) were purchased from Sigma. Caspase inhibitor benzyloxycarbonylvaline-alanine-asparate-fluoromethylketone (Z-VAD.fmk) was purchased from Bachem (King of Prussia, PA). The TUNEL kit APO-BRDU was obtained from Phoenix Flow Systems (San Diego, CA).
Induction of Apoptosis and Necrosis
The MDA-468 breast cancer cell line was maintained as a monolayer culture in IMEM medium supplemented with 5% FBS. Apoptosis was induced by the addition of 0.5 μM staurosporine to confluent cultures. Attached cells were removed by trypsin–EDTA 3 hr after addition of the drug and floating cells were collected after 5 hr of drug incubation. Suspension cultures of the MOLT-4 cell line and its drug-resistant subline MOLT-4/R9 (Frankfurt et al. 1994) were grown in RPMI 1640 medium supplemented with 10% FBS, 2 mM glutamine and 10–5 M 2-mercaptoethanol. Apoptosis was induced in cultures at a density of 0.7–1.0 × 106 cells/ml with 0.25–0.5 μM staurosporine.
For induction of necrosis, MDA-468 cells were removed with trypsin–EDTA and resuspended in medium supplemented with 5% FBS. Cell suspensions were treated with 0.5 M sodium azide for 3 hr at 37C or with 0.02% saponin in PBS for 1 hr at 37C. For induction of cell death by hyperthermia, cell suspensions were heated at 56C for 1 hr and incubated at 37C for 1 hr. Single-stranded DNA breaks were induced by treatment of the monolayer cultures with 50 mM H2O2 for 10 min. The presence of DNA breaks was confirmed by alkaline denaturation using a microplate assay for DNA damage determination (Batel et al. 1999)
DNA Denaturation and Staining with the MAb Against ssDNA
Cells were fixed in methanol–PBS (6:1) for 24–72 hr (Frankfurt et al. 1996). Fixed cells were resuspended in 0.25 ml of formamide (5 × 105 cells in 15-ml plastic tubes) and heated in a water bath at 37–90C for 10 min. After heating, cells were washed with 2 ml of 1% non-fat dry milk in PBS, resuspended in 100 μl of MAb F7–26 (10 μg/ml in PBS containing 5% FBS), and incubated for 15 min. Cell were then rinsed with PBS and stained with fluorescein-conjugated anti-mouse IgM (1:50 in PBS containing 1% non-fat dry milk) for 15 min. For flow cytometry, cells were rinsed with PBS and resuspended in 0.5 ml of PBS containing 1 μg/ml propidium iodide. For fluorescence microscopy, cytocentrifuge preparations of antibody-stained cells were counter-stained with 0.1 μg/ml DAPI in PBS for 10 min. Treatment of cells with S1 nuclease was performed as described earlier (Frankfurt 1994; Frankfurt et al. 1996). All data are presented as mean ± SE for at least four experiments.
TUNEL Assay
Staining with the APO-BRDU kit was performed according to the manufacturer's instructions. Cells were fixed in ice-cold 1% paraformaldehyde for 15 min, washed with PBS, and fixed in 70% ethanol at −20C for 24 hr. Cells were then incubated in buffer containing terminal deoxynucleotidyl transferase and bromodeoxyuridine triphosphate for 3 hr at 37C, stained with fluorescein-conjugated anti-BrDU antibody for 30 min, and counterstained with propidium iodide.
Flow Cytometry
Cell suspensions stained with MAb and TUNEL were analyzed on a FACScan flow cytometer (Becton–Dickinson; Sunnyvale, CA). Green fluorescence of fluorescein-labeled antibodies and red fluorescence of the propidium iodide bound to DNA was measured for 5,000–10,000 cells. Distributions of green fluorescence were generated after elimination of debris and cell clumps in two-parameter dot-plots using DNA/propidium iodide fluorescence for gating. Mean fluorescence intensity of DNA bound antibody was calculated with the LYSIS II program (Becton–Dickinson). DNA histograms were generated by flow cytometric analysis of fixed cells stained with 50 μg/ml propidium iodide and 20 μg/ml RNase in PBS.
Fluorescence Microscopy
Cytocentrifuge preparations of cells stained with MAb F7–26 and DAPI were observed under an Olympus BH-2 fluorescence microscope using UV excitation for DNA fluorochrome DAPI and 450–490-nm excitation for fluorescein-labeled antibody. To determine the relation between chromatin morphology and antibody binding, the same field was photographed with UV and visible light excitation.
Staining of Tissue Sections
Female Balb/c mice were injected IP with 500 mg/kg hydroxyurea or 250 mg/kg hydrocortisone. Thymus and small intestine were removed 4 hr later and processed with three different protocols.
Protocol 1: Paraffin Sections. Tissues were fixed in 10% buffered formalin at 4C for 24 hr and embedded in paraffin. Sections (4 μm) were deparaffinized and treated with PBS containing 0.2 mg/ml saponin and 20 μg/ml proteinase K for 20 min. Sections were then rinsed with distilled water and heated in 50% formamide prewarmed to 56C for 20 min. Positive staining of apoptotic cells was obtained in the temperature range of 37–65C, but the formamide concentration had to be precisely 50% and could not be varied. After heating, slides were transferred into ice-cold PBS, blocked in 3% non-fat dry milk, and stained with MAb F7–26 (10 μg/ml PBS containing 5% FBS) and peroxidase-conjugated anti-mouse IgM (1:100 in PBS). DAB was used as a chromogen and Hill's hematoxylin as a counterstain.
Protocol 2: Frozen Sections From Tissues Fixed in Cold 4% Paraformaldehyde for 24 Hr. Slides with sections were treated with methanol: PBS (6:1) for 30 min, and air-dried. Slides with dried sections were transferred into saponin–proteinase K solution and treated as described above for paraffin sections.
Protocol 3: Frozen Sections from Fresh Tissues. Sections were fixed in 4% paraformaldehyde for 10 min, rinsed in PBS, treated with methanol: PBS (6:1), and air-dried. Slides with dried sections were treated in PBS containing 0.2 mg/ml saponin (without proteinase K), heated in formamide, and stained as described above for paraffin sections.
Results
DNA Denaturation by Formamide in Apoptotic Cells
In cultures of breast cancer MDA-468 cells treated with the protein kinase inhibitor staurosporine, only cells with chromatin condensation typical of apoptosis were labeled with antibody against ssDNA. In contrast, cells with dispersed chromatin in staurosporine-treated cultures or interphase nuclei of untreated cultures did not react with the anti-ssDNA MAb (Figures 1A and 1B). Intense antibody fluorescence was observed in cells with early stages of chromatin margin-ation 3 hr after addition of the drug. Bright fluorescence was also seen in fragmented nuclei of floating late apoptotic cells after 5 hr of drug treatment. Membrane integrity was retained in 99% of the attached and the floating cells, as demonstrated by the trypan blue dye exclusion. Correlation between chromatin condensation typical of apoptosis and formamide-induced binding of the anti-ssDNA MAb was also observed in the staurosporine-treated leukemic MOLT-4 cell line and its drug-resistant subline MOLT-4/R (Figures 1C and 1D). Staining of condensed and marginated chromatin in MOLT-4/R cells is especially interesting because apoptosis is not accompanied by DNA fragmentation in this cell line (Frankfurt et al. 1994, 1996) (see also below).
To determine whether formamide-induced DNA denaturation is specific for condensed apoptotic chromatin, actively proliferating MDA-468 and MOLT-4 cells were heated in formamide and stained with MAb F7–26. Fifty mitotic figures were counted in each sample and no MAb labeling was detected. These data show that specific changes in condensed apoptotic chromatin are responsible for the increased sensitivity of DNA to formamide, whereas chromatin condensation in the mitotic cells has no effect on DNA sensitivity to formamide.
Flow Cytometry of the Apoptotic and Necrotic Cells
The ability of the formamide–MAb technique to distinguish between apoptosis and necrosis was determined by flow cytometric analysis of cells treated with apoptosis- and necrosis-inducing agents. Necrosis was induced in MDA-468 cells with the respiratory chain inhibitor sodium azide (Lizard et al. 1995), the membrane-damaging detergent saponin (Dong et al. 1997), or hyperthermia (Lennon et al. 1991). The necrotic mechanism of cell death after these treatments was confirmed by the loss of membrane integrity (trypan blue staining in 100% of cells) and by the absence of chromatin condensation or nuclear fragmentation.
Fluorescence profiles of cells heated in the presence of formamide and stained with the anti-ssDNA MAb are shown in Figure 2. Two subsets of cells were observed after 3-hr treatment with staurosporine: cells with low fluorescence, similar to that of the untreated cells, and brightly fluorescent apoptotic cells. Ninety percent of cells were detached from the substratum 5 hr after the addition of staurosporine, and all these cells were intensely stained with the anti-ssDNA MAb. Fluorescence microscopy demonstrated that all floating cells had chromatin condensation and nuclear fragmentation typical of apoptosis. In contrast to the high fluorescence intensity of the apoptotic cells, the necrotic cells in all three models of necrosis had a fluorescence profile similar to that of untreated control cells (Figure 2). These data were confirmed by fluorescence microscopy, which demonstrated only unstained necrotic cells.
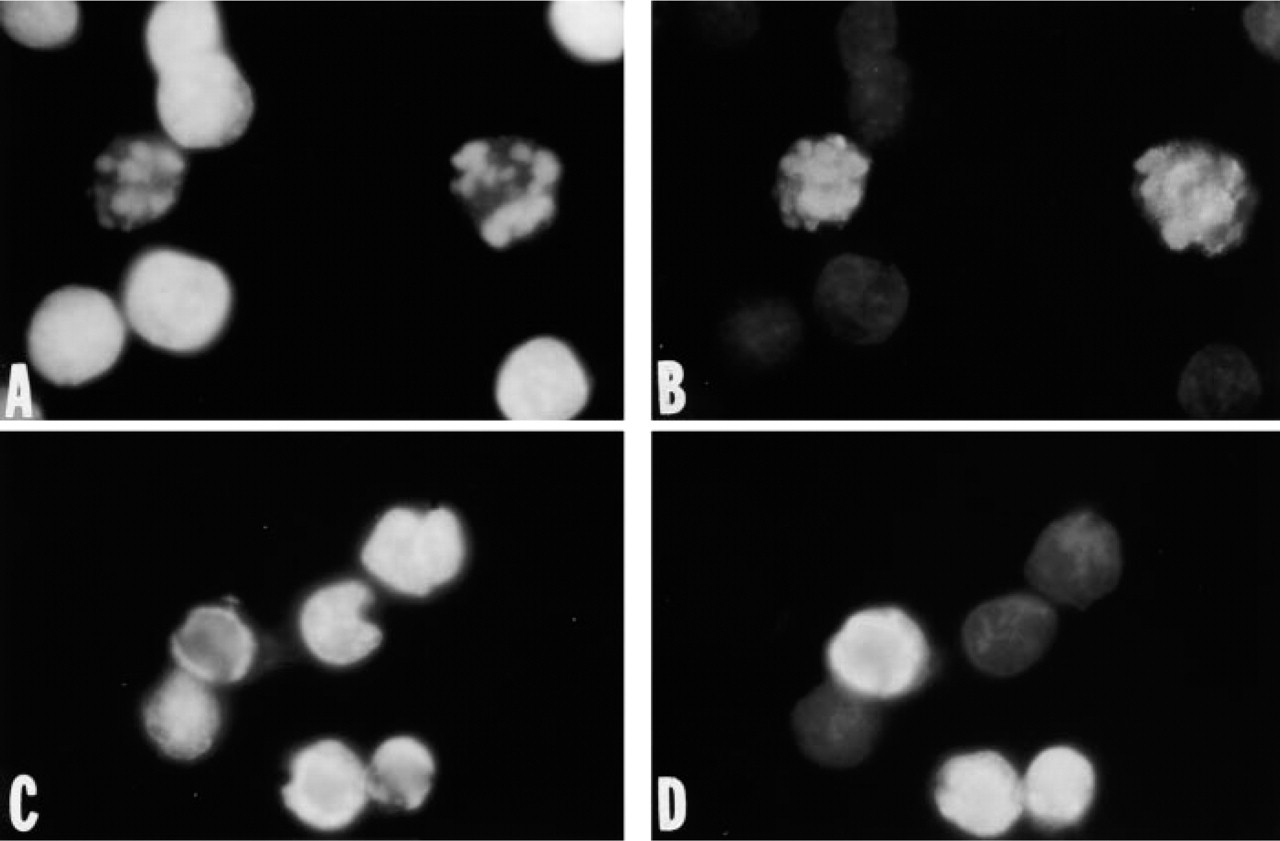
Staining of condensed chromatin in apoptotic nuclei with an MAb against ssDNA. Fluorescence microphotographs of MDA-468 (
To confirm the specificity of MAb F7–26 binding to ssDNA, cells heated in formamide were treated with 100 U/ml of S1 nuclease to digest DNA in single-stranded conformation. This treatment completely eliminated the subset of intensely fluorescent apoptotic cells in staurosporine-treated cultures and had no effect on the fluorescence of the non-apoptotic cells.
Temperature Kinetics of DNA Denaturation
The observations described above were made on cells heated at 75C. For a detailed analysis of formamide effect on DNA stability, apoptotic and necrotic cells were heated at 37–90C and stained with the anti-ssDNA MAb. To study the effect of ssDNA breaks, cells treated with H2O2 were used. Fluorescence of apoptotic cells progressively increased after the heating of tubes at 50–90C (Figure 3). The heating at 75–80C induced the maximal difference in fluorescence of the apoptotic and the non-apoptotic cells.
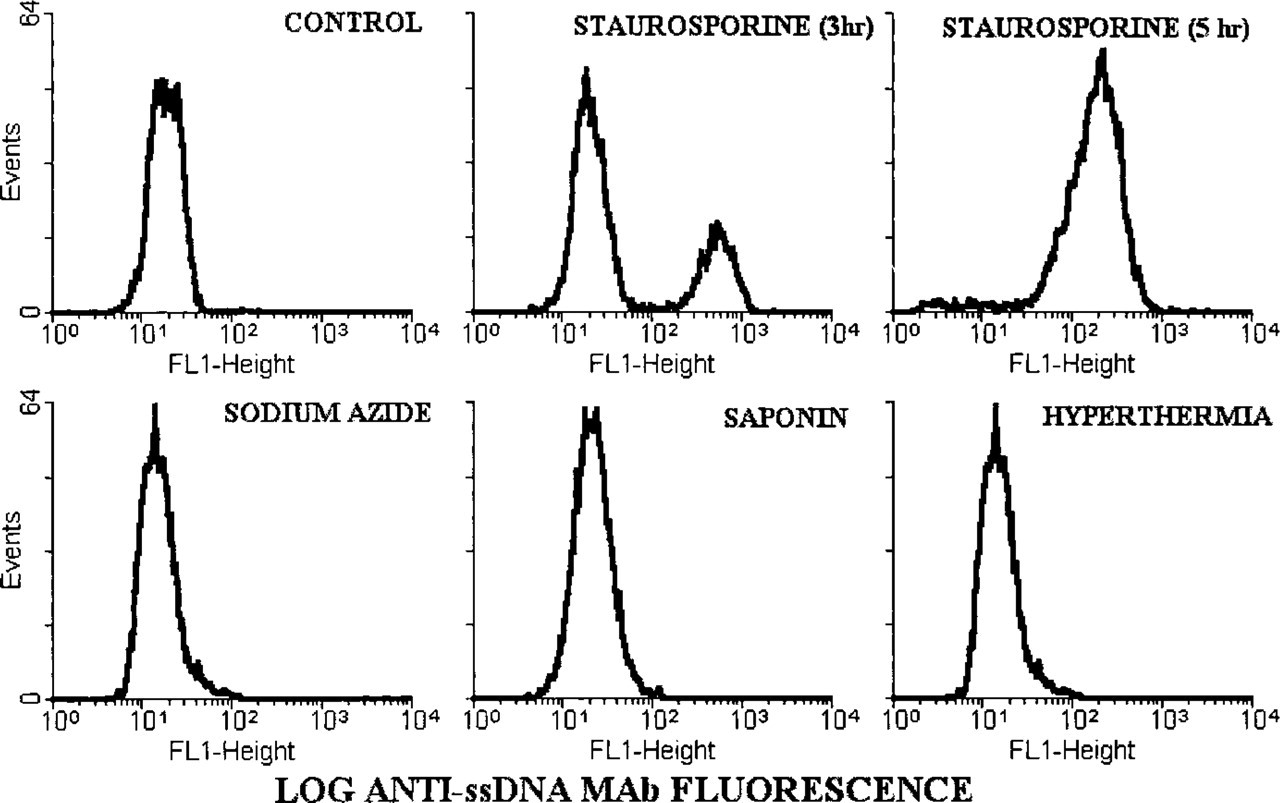
MAb to ssDNA staining of apoptotic but not of necrotic cells. Fluorescence distributions of MDA-468 cells heated in formamide and stained with MAb F7–26 were generated on a flow cytometer. Apoptosis was induced by staurosporine and necrosis was induced by sodium azide, saponin, or hyperthermia. Note that apoptotic cells are intensely stained with the MAb, whereas the fluorescence profiles of necrotic and control cells are similar.
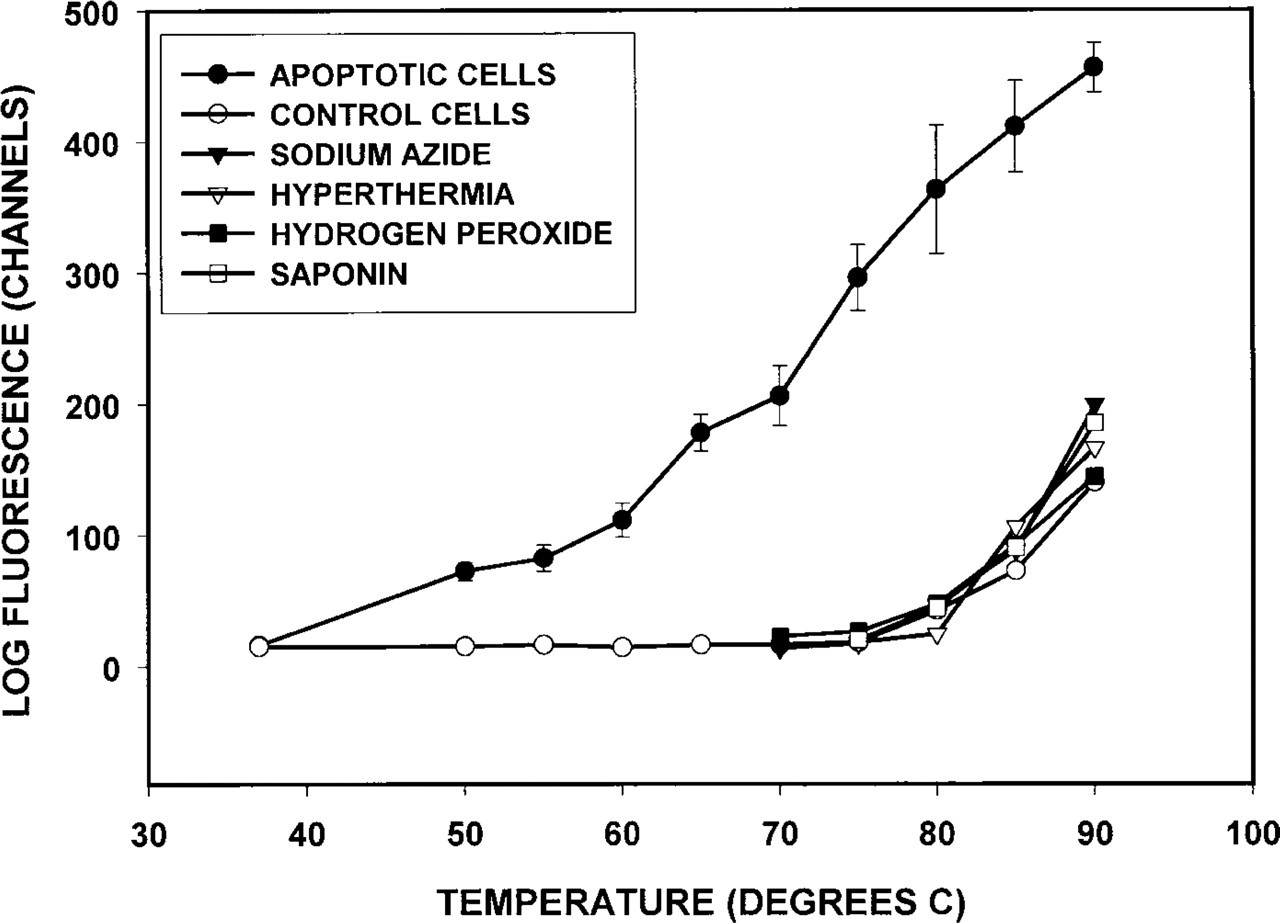
Temperature kinetics of DNA denaturation in MDA-468 cells. Cells were heated in formamide at various temperatures for 10 min and stained with MAb F7–26. Apoptosis was induced by treatment with staurosporine for 3 hr. Necrosis was induced by sodium azide, saponin, or hyperthermia. Single-stranded DNA breaks were induced by H2O2. Mean fluorescence intensity of DNA bound antibody was measured on a flow cytometer. Note that after heating at 56–80C only apoptotic cells bind the antibody.
The fluorescence of untreated, necrotic and H2O2-treated cells did not change significantly after heating at 37–80C and increased only after heating at 85–90C (Figure 3). However, even after heating at 90C, the fluorescence of MAb-stained necrotic and H2O2-treated cells was similar to that of the untreated cells. Therefore, in the presence of formamide the DNA sensitivity to thermal denaturation was increased in the apoptotic cells but not in the necrotic cells or in the cells with DNA breaks.
TUNEL Staining of Apoptotic and Necrotic Cells
Labeling of 3′-hydroxyl ends in DNA by TUNEL was performed to compare the sensitivity of the two techniques and to determine whether necrotic cells, which did not bind the anti-ssDNA MAb, did have double-stranded DNA breaks. Early and late apoptotic cells in cultures treated with staurosporine for 3 and 5 hr were stained by TUNEL (Figure 4). However, staining of early apoptotic cells by TUNEL was significantly weaker than staining of the late apoptotic cells (mean fluorescence intensity 160 ± 11 and 294 ±33 channels, respectively; n = 4; p = 0.01). This is in contrast to the higher fluorescence of the early than of the late apoptotic cells with the formamide–MAb method (mean fluorescence 359 ± 28 and 219 ± 40 channels, respectively; n = 6; p = 0.03).
The most important observation was the difference in the staining of necrotic cells by the two procedures. TUNEL stained 90–100% of necrotic cells in cultures treated with sodium azide, saponin, or hyperthermia, with staining intensity similar to that of the late apoptotic cells (Figure 4). In contrast, necrotic cells in all three models of necrosis did not stain with the anti-ssDNA MAb (Figure 2).
Caspase Inhibition and MAb Staining
Treatment with a caspase inhibitor was performed to determine whether prevention of chromatin condensation would inhibit MAb staining. Addition of 200 μM of the pan-caspase inhibitor Z-VAD.fmk to staurosporine-treated MDA-468 cultures completely prevented chromatin condensation and MAb staining. In cultures treated for 3 hr with staurosporine alone, 39 ± 2% of cells were apoptotic and stained with the MAb, whereas in cultures treated with both drugs, all cells remained attached, had normal chromatin morphology, and did not bind the anti-ssDNA MAb. Almost complete inhibition of apoptosis by the caspase inhibitor was obvious even after 18 hr of incubation. In cultures treated with staurosporine alone for 18 hr, all cells were floating, had condensed chromatin, and showed intense antibody fluorescence. In cultures treated with staurosporine and the caspase inhibitor, 90% of cells remained attached, had dispersed chromatin, and did not stain with the MAb. Inhibition of apoptosis in this model was not accompanied by the development of secondary necrosis, as demonstrated by the absence of membrane permeability to trypan blue.
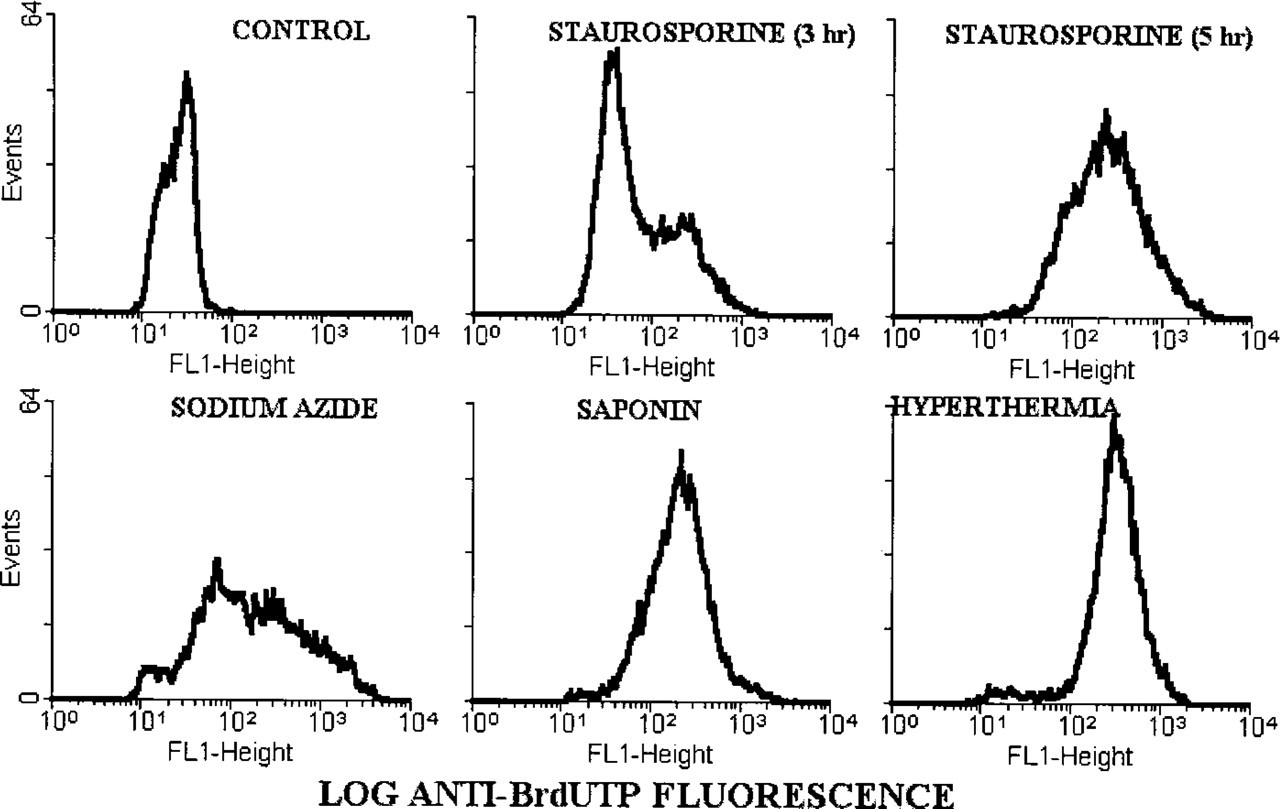
TUNEL staining of apoptotic and necrotic cells. Fluorescence distributions of MDA-468 cells stained with TUNEL were generated on a flow cytometer. Apoptosis was induced by staurosporine and necrosis was induced by sodium azide, saponin, or hyperthermia. Note that cells at early stage of apoptosis (staurosporine 3 hr) are weakly stained, whereas late apoptotic cells (staurosporine 5 hr) and necrotic cells are intensely stained by TUNEL.
Staining in the Absence of DNA Fragmentation
MOLT-4/R9 cells were used to determine whether the formamide–MAb procedure could detect apoptosis in the absence of DNA fragmentation. Apoptosis in MOLT-4/R9 cells is not accompanied by low molecular weight DNA fragmentation, in contrast to the parent cell line (Frankfurt et al. 1996). Fluorescence histograms demonstrated that in MOLT-4 cultures treated with 0.25 μM staurosporine, 51 ± 2% of cells were apoptotic, while in MOLT-4/R9 cultures treated with 0.5 μM of staurosporine apoptosis was seen in 40 ± 1% of cells. Fluorescence intensity of the positive cells was similar in the two cultures (mean fluorescence 477 ± 22 and 465 ± 21 channels, respectively).
In contrast to the similarity in fluorescence distributions, there was a significant difference in DNA histograms of the two cell lines treated with staurosporine (Figure 5). In MOLT-4 cells, apoptosis was accompanied by DNA fragmentation as demonstrated by the presence of cells with hypodiploid DNA content (47 ± 6% of total), whereas in the resistant subline only a small shoulder was detected (3% of cells with slightly decreased DNA content). Fluorescence microscopy demonstrated chromatin condensation and margination typical of apoptosis in all MOLT-4/R9 cells stained with the anti-ssDNA MAb (Figures 1C and 1D).
Detection of Apoptosis in Tissue Sections
We used two in vivo models of drug-induced apoptosis to identify apoptotic cells by the formamide–MAb technique in tissue sections. Apoptosis was induced in the crypts of mouse small intestine with the DNA synthesis inhibitor hydroxyurea and in the cortex of mouse thymus with hydrocortisone (Sarraf et al. 1993; Nkamura et al. 1997). Sections of paraffin-embedded tissues and frozen sections were heated in formamide and stained with the anti-ssDNA MAb.
Staining of formalin-fixed, paraffin-embedded tissue sections is illustrated in Figure 6. Apoptotic cells with condensed chromatin and fragmented nuclei in the crypts of the small intestine from hydroxyurea-treated mice reacted positively with the MAb (Figure 6A). No reactivity was seen in most crypts of untreated animals (Figure 6B). Only 5–10% of crypts contained single isolated positive cells, in agreement with the known low frequency of apoptosis in untreated animals (Merritt et al. 1994). Multiple foci of MAb-positive apoptotic cells were detected in the thymus of mice treated with the glucocorticoid, whereas only rare stained cells were seen in the thymus of the untreated animals (Figures 6C and 6D). There was no staining in control sections from drug-treated tissues when heating in formamide was omitted or when the anti-ssDNA MAb was replaced by the antibody solvent. Staining was weak and most apoptotic cells were negative in sections not treated with proteinase K.
Staining of frozen sections from thymuses of hydrocortisone-treated and untreated mice produced results similar to those of paraffin sections (Figures 6C and 6D). Pretreatment with proteinase K was not necessary for positive staining of apoptotic cells when frozen sections from fresh tissues were fixed in paraformaldehyde for 10 min. However, frozen sections prepared from tissues fixed in paraformaldehyde for 24 hr stained only if pretreatment with proteinase K was included in the protocol. Therefore, treatment with proteinase K was necessary for formamide–MAb staining only after prolonged fixation. In the absence of formalin fixation or after 10-min fixation, positive staining of apoptotic cells was obtained without proteinase K pretreatment.
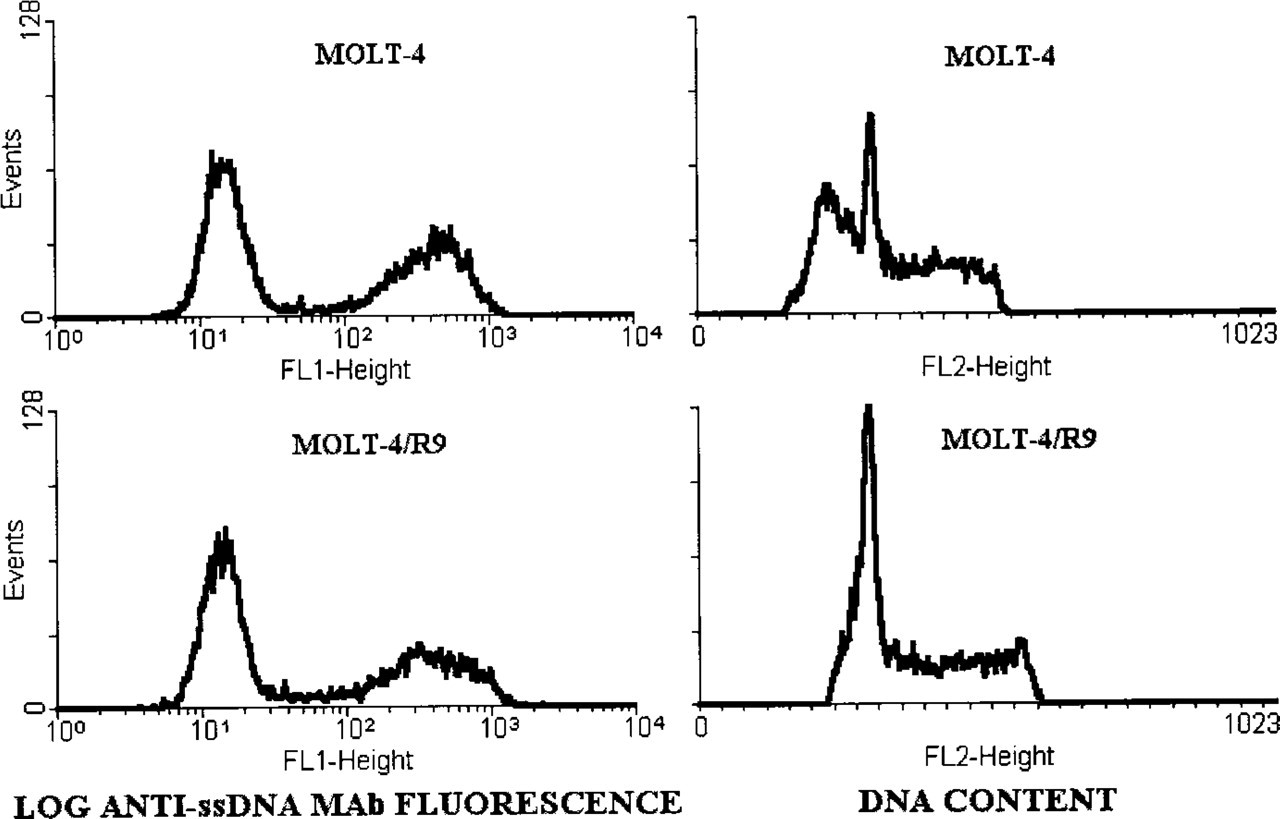
DNA fragmentation and the staining of apoptotic cells with the MAb against ssDNA. Fluorescence profiles and DNA histograms of MOLT-4 and MOLT-4/R9 cells treated with staurosporine for 3 hr are shown. Treatment induced apoptosis in 50% of MOLT-4 cells and in 40% of MOLT-4/R9 cells. Note that the anti-ssDNA MAb stains apoptotic cells in both cultures with a similar intensity (mean fluorescence 477 and 465 channels), while DNA distributions demonstrate the presence of hypodiploid cells with fragmented DNA only in MOLT-4 cultures.
Paraffin sections from thymuses and small intestine of untreated mice were treated with 1 μg/ml DNAse before heating in formamide and staining with the anti-ssDNA MAb. Such treatment induces double-stranded DNA breaks and is used as a standard positive control for the TUNEL procedure (Gavrieli et al. 1992; Frankfurt et al. 1997). However, in contrast to TUNEL, DNase did not induce staining of non-apoptotic cells by the anti-ssDNA MAb. These data demonstrate that staining of apoptotic cells with the formamide–MAb technique is not based on the detection of DNA breaks.
Discussion
The data from the present study show that increased sensitivity to thermal denaturation in condensed chromatin of apoptotic nuclei can be used for specific and probably universal detection of apoptotic cells. A novel finding in this study is the selective denaturation of DNA in apoptotic cells in the presence of formamide. Denatured DNA in condensed chromatin of the apoptotic nuclei was detected with MAb F7–26, which specifically reacts with deoxycytidine and requires for its binding ssDNA of at least 25–30 bases in length (Frankfurt 1990; Frankfurt et al. 1996). As shown in three different models, this MAb does not react with the necrotic cells.
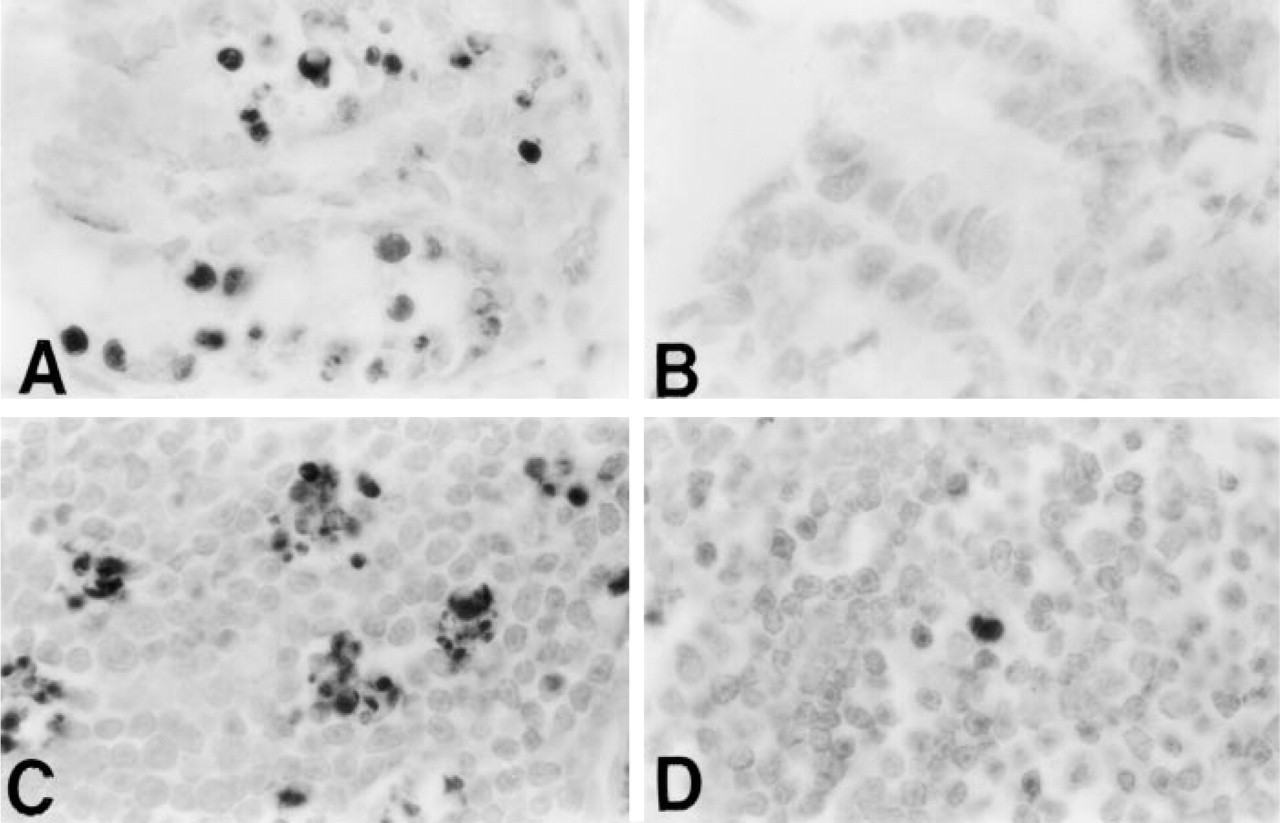
Staining of apoptotic cells in tissue sections with the MAb against ssDNA. Photomicrographs of small intestine crypts of a mouse treated with hydroxyurea (
The following binding characteristics of the anti-ssDNA MAbs are critical for their ability to specifically stain the apoptotic cells: absence of any crossreactivity with native double-stranded DNA and large size of the binding site. MAbs F7–26 and AP-13 with these binding characteristics were used in our previous studies to detect ssDNA in situ (Frankfurt 1990; Frankfurt et al. 1997). Because a relatively large stretch of DNA in single-stranded conformation is required for the binding, these antibodies do not react with single-stranded DNA ends. This reactivity is responsible for the nonspecificity of the TUNEL assay, because ssDNA ends are present in the necrotic cells.
The specific reactivity of the MAb with apoptotic DNA is induced by formamide, known as a gentle DNA denaturing agent (Marmur and Ts'o 1961; Levine et al. 1963; Blake and Delcourt 1996). The high sensitivity to thermal denaturation of DNA in condensed chromatin of apoptotic cells is sufficient to produce large stretches of ssDNA in the presence of formamide at low temperature. In contrast, strong denaturing agents, such as hydrochloric acid, induce DNA denaturation in apoptotic and necrotic cells and are not suitable for the specific staining of apoptotic cells (Hotz et al. 1992).
Polyclonal rabbit antibodies against ssDNA were used to detect apoptotic cells in tissue sections (Naruse et al. 1994; Umemura et al. 1996; Watanabe et al. 1999). These antibodies bind to ssDNA stretches 3 bases long and stain apoptotic cells without heating by detecting single-strand ends. In contrast, significantly longer stretches of ssDNA are needed for the binding of MAb F7–26 used in our studies. This MAb does not react with DNA ends and detects ssDNA induced by formamide denaturation. Thus, detection of apoptotic cells by two types of anti-ssDNA antibodies is based on different principles. Whereas polyclonal antibody detects free DNA ends nonspecific for apoptosis, MAb F7–26 binding reflects specific changes in condensed apoptotic chromatin. Another technique used to detect ssDNA in apoptotic cells is based on the ability of acridine orange to distinguish between DNA in single- and double-stranded conformation (Dyson et al. 1986; Hotz et al. 1992).
The formamide–MAb technique described here has significant advantages compared to other methods used to detect ssDNA in apoptotic cells: (a) Specificity: polyclonal anti-ssDNA antibodies stained nonspecific necrotic foci (Watanabe et al. 1999), while the hydrochloric acid–acridine orange technique detected ssDNA in apoptotic, necrotic, and normal mitotic cells (Hotz et al. 1992). In contrast, MAb F7–26 specifically stains apoptotic cells and has no reactivity with the necrotic and the mitotic cells. (b) Sensitivity: The formamide–MAb method is more sensitive than TUNEL, especially for early stages of apoptosis. In contrast, the apoptotic index measured by polyclonal antibodies was threefold lower than the TUNEL index (Watanabe et al. 1999). (c) Application to tissue sections: Acridine orange obviously could not be used for histological staining, whereas the formamide–MAb technique is easily applied for routine staining of tissue sections.
Formamide-induced DNA denaturation is also a major improvement compared to our previous method based on intense heating in the absence of formamide (Frankfurt et al. 1996, 1997). First, the formamide–MAb technique can be applied for staining of frozen sections and sections from formalin-fixed, paraffin-embedded tissues, whereas DNA denaturation by intense heating in the absence of formamide was inhibited by formalin-induced DNA–protein crosslinking and was not applicable to frozen sections. Second, although the sensitivity of both methods is similar, the specificity is more easily achieved by the formamide technique. Heating in the absence of formamide produced specific staining under relatively narrow conditions, such as specific MgCl2 concentration (Frankfurt 1994), whereas only apoptotic DNA denatured in the presence of formamide.
It was important to determine whether increased sensitivity of the apoptotic DNA to thermal denaturation in the presence of formamide is caused by DNA breaks. The role of DNA breaks is critical for the assessment of specificity of the technique because the current view is that double-stranded DNA breaks are not specific for apoptosis (Collins et al. 1992; Fukuda et al. 1993; Dong et al. 1997; Kanoh et al. 1999).
The following facts demonstrate that selective denaturation of DNA of the apoptotic nuclei in the presence of formamide may not be related to DNA breaks. (a) Necrotic cells with high levels of double-stranded DNA breaks demonstrated by TUNEL did not react with the anti-ssDNA MAb. (b) Apoptotic cells with very low and high levels of DNA fragmentation were stained by the formamide–MAb technique with similar intensity. (c) Nuclei with single-stranded DNA breaks induced by H2O2 treatment did not bind the MAb after heating in formamide, although alkaline denaturation detected DNA breaks in these nuclei. (d) Induction of double-stranded DNA breaks by DNase did not produce MAb positivity, although this treatment induces TUNEL staining.
These data indicate that DNA denaturation in the apoptotic cells in the presence of formamide is induced by changes in chromatin associated with condensation and not influenced by DNA breaks. The occurrence of chromatin condensation typical of apoptosis in the absence of DNA fragmentation is in agreement with this conclusion (Cohen et al. 1992; Oberhammer et al. 1993; Ormerod et al. 1994; Allera et al. 1997; Sakahira et al. 1999). The fact that formamide decreased DNA stability in condensed chromatin of apoptotic cells but had no such effect on the condensed chromatin of mitotic nuclei may be explained by different mechanisms involved in the two types of condensation. Indeed, phosphorylation of histones is required for mitotic condensation, whereas deacetylation of histones is associated with apoptotic condensation (Allera et al. 1997).
Because the effect of formamide was not related to the presence of DNA breaks, it is important to understand what other factors may be responsible for the high sensitivity of the apoptotic DNA to denaturation. We suggest that the changes in DNA–histone interactions in condensed chromatin of apoptotic nuclei may be responsible, at least partially, for the high sensitivity to formamide. This hypothesis is supported by the fact that histones stabilize DNA against thermal denaturation (Tsai et al. 1975). Digestion of histones during apoptosis may decrease the stability of DNA (Kaufmann 1989; Kuribayashi et al. 1996). Decreased interaction of histones with apoptotic DNA is indicated by the increased immunoreactivity and extract-ability of histones during early apoptosis before the appearance of DNA breaks (Zunino et al. 1996).
In conclusion, heating of fixed cells or tissue sections in the presence of formamide induces DNA denaturation in apoptotic but not in necrotic cells. Staining of denatured DNA with the MAb specific for ssDNA makes possible detection of the apoptotic cells and discrimination of apoptosis from necrosis. Increased sensitivity of DNA to thermal denaturation is associated with specific changes in the condensed chromatin and is not induced by single- or double-stranded DNA breaks. The formamide–MAb method detects apoptotic cells in tissues processed with routine histological techniques.
Footnotes
Acknowledgements
Supported by NIH Grant CA83508.