
Abstract
We developed an isolation technique for motor neurons from adult rat spinal cord. Spinal cord enlargements were discretely microdissected into ventral horn tissue columns that were trypsin-digested and subjected to differential low-speed centrifugation to fractionate ventral horn cell types. A fraction enriched in α-motor neurons was isolated. Motor neuron enrichment was verified by immunofluorescence for choline acetyltransferase and prelabeling axon projections to skeletal muscle. Adult motor neurons were isolated from naïve rats and were exposed to oxidative agents or were isolated from rats with sciatic nerve lesions (avulsions). We tested the hypothesis, using single-cell gel electrophoresis (comet assay), that hydrogen peroxide, nitric oxide, and peroxynitrite exposure in vitro and axotomy in vivo induce DNA damage in adult motor neurons early during their degeneration. This study contributes three important developments in the study of motor neurons. It demonstrates that mature spinal motor neurons can be isolated and used for in vitro models of motor neuron degeneration. It shows that adult motor neurons can be isolated from in vivo models of motor neuron degeneration and evaluated on a single-cell basis. This study also demonstrates that the comet assay is a feasible method for measuring DNA damage in individual motor neurons. Using these methods, we conclude that motor neurons undergoing oxidative stress from reactive oxygen species and axotomy accumulate DNA damage early in their degeneration. (J Histochem Cytochem 49:957–972, 2001)
Keywords
G
A variety of endogenous or intrinsic and exogenous or environmental factors can cause DNA damage (Lindahl 1993; Subba Rao 1993). Endogenous sources include reactive oxygen species (ROS) and oxidative stress generated by cellular metabolism, temperature, errors in DNA replication and repair, high levels of glucose and other reducing sugars, and methylation. Intrinsically generated DNA damage occurs as mismatched base pairs, base structure alterations such as tautomeric shifts and deamination, base adducts (e.g., hydroxylation), and base deletions causing apurinic/apyrimidinic (AP) sites (alkali-labile sites), single-strand breaks (SSBs), and double-strand breaks (DSBs). Estimates indicate that a human cell sustains ∼10,000 lesions per day due to metabolism-generated free radicals (Subba Rao 1993) and 2000–10,000 DNA purine bases turn over daily because of hydrolytic depurination and subsequent repair (Lindahl 1993). Environmental sources of DNA damage include dietary mutagenic chemicals, ultraviolet and ionizing radiation such as X-rays and γ-rays, free radicals, and heavy metals. These exogenous agents can cause crosslinks, adducts, and oxidative cleavage. Many of these types of DNA damage can be converted from one form to another form. For example, the interaction of DNA with hydroxyl radicals yields a variety of lesions, including base adducts, strand breaks, and sugar modifications; in addition, AP sites are converted into SSBs if not repaired.
DNA lesions can be evaluated by a variety of methods. Some methods are quantitative and other methods are qualitative. The methods for DNA damage detection can be broadly classified as biochemical and in situ detection methods. DNA filter elution assay is a powerful biochemical method for the determination of DNA strand breaks, apurinic/apyrimidinic (alkalilabile) sites, and crosslinks (Kohn 1991). Another method is the detection of nucleoside adducts in digests of DNA extracts with high-performance liquid chromatography (Ames 1989). Gel electrophoresis is another reliable and major means for detecting genomic DNA damage (i.e., DNA fragmentation) in tissue/cell extracts. Flow cytometry for sorting cells on the basis of DNA quantity, indicating DNA damage, is also a good method for quantification of numbers of cells with DNA damage and severity of DNA damage. The most popular in situ method for DNA damage is the terminal deoxynucleotidyl transferase-mediated 3′ dUTP nick end-labeling (TUNEL) technique (Gavrieli et al. 1992). Neuroscientists often use this method to observe cell death in nervous system disease and injury.
We are interested in developing sensitive and quantitative assays for detecting genotoxicity directly in motor neurons because of its relevance to ALS (Martin 2000; Martin et al. 2000). Single-cell gel electrophoresis, also called the “comet” assay, is a method for identifying early damage to genomic DNA of eukaryotic cells on a single-cell basis (Östling and Johanson 1984; Singh et al. 1988; Kindzelskii and Petty 1999; Morris et al. 1999; Tice et al. 2000). The types of DNA lesions detectable by comet assay include SSBs, alkali-labile sites (AP sites), and DSBs. Based on the principles of alkaline elution, the comet assay detects early DNA damage, which is of particular importance in investigating the mechanisms of cell death, particularly apoptosis. SSBs and DSBs are strong signals for activation of p53-mediated cell death (Jayaraman and Prives 1995). p53 is activated or induced in cells undergoing DNA damage-induced apoptosis. This pathway may participate in the mechanisms of motor neuron degeneration in ALS (Martin 2000). The goals of these experiments were first, to develop a new method to isolate and study adult motor neurons and, second, to apply the comet assay to study of DNA damage in motor neurons.
Materials and Methods
Animals and Tissues
Adult male Sprague-Dawley rats (Charles River; Wilmington, MA) weighing ∼150–200 g were used for these experiments. Naïve rats without experimental manipulations and rats with experimental manipulations were used in these experiments. The manipulations performed were retrograde tracing of motor neurons and avulsion of the sciatic nerve. The institutional Animal Care and Use Committee approved the animal protocols. The animals were housed in a colony room with a 12 hr:12 hr light:dark cycle and ad libitum access to food and water.
Retrograde Tracing of Spinal Motor Neurons
Rats (n = 2) were anesthetized deeply with enflurane:oxygen:nitrous oxide (1:33:66) and, using sterile surgery, the sciatic nerve was exposed within the middle of the upper hindlimb. The nerve was transected by cutting and 50 μl of 2.5% DAPI was applied to the proximal nerve stump for 1 hr using tracer-saturated Gelfoam placed in a sterile Eppendorf tube. The incision was closed with the bottom of the Eppendorf tube containing DAPI remaining applied to the proximal stump of the sciatic nerve. The animals were allowed to survive for 48 hr before they were sacrificed.
Sciatic Nerve Avulsion Model of Motor Neuron Apoptosis
The unilateral sciatic nerve avulsion model was used as an in vivo model of spinal motor neuron apoptosis (Martin et al. 1999; Liu and Martin 2001). Rats were anesthetized deeply with enflurane:oxygen:nitrous oxide (1:33:66). With sterile surgery, a midline incision was made in the lateral aspect of the left pelvis and upper hindlimb. The sciatic nerve was located by blunt retraction of the biceps femoris and gluteus muscles and was tracked proximally to an extravertebral location deep within the pelvis. A steady, moderate traction was applied to the sciatic nerve with forceps until the nerve separated from the spinal cord, resulting in a mixed motor-sensory root avulsion. Muscle retraction was released and the overlying skin was sutured. The animals (n = 2–4 rats per time point) were allowed to live for 5, 7, 10, 14, or 28 days after sciatic nerve avulsion.
Preparation of Spinal Cord Tissue for Isolation of Mature Motor Neurons
Spinal cords were isolated from adult rats that were anesthetized deeply with a mixture of enflurane:oxygen:nitrous oxide (1:33:66) and then decapitated. From rats without experimental lesions, cervical and lumbar enlargements were used. The two enlargements of the spinal cord contain the majority of the spinal motor neurons. From rats exposed to tracers and from rats with sciatic nerve avulsions, only the entire lumbar enlargements (divided into ipsilateral and contralateral sides) were used. After removal of the pia, lumbar/cervical enlargements were dissected segmentally under a surgical microscope and then the segments were microdis-sected into gray matter columns of ventral horn without appreciable contamination of dorsal horn and surrounding white matter funiculi. Spinal motor neurons are large neurons locating in Lamina IX of spinal cord. Gray matter tissue columns from spinal cord ventral horns of lumbar/cervical enlargements were collected and rinsed in a cell culture dish on ice containing dissection medium [1 × Ca2+ and Mg2+-free Hanks balanced salt solution (Gibco BRL; Grand Island, NY) supplemented with glucose and sucrose]. These tissues were used to prepare motor neuron cell suspensions.
Preparation of Adult Spinal Motor Neuron-enriched Cell Suspensions
Digestion of Spinal Cord Ventral Horns. Ventral horn samples were digested (20 min) with 0.25% trypsin-EDTA (Gibco) in a tissue culture incubator (5% CO2 and 95% air at 37C). This mixture was titurated gently with a transfer pipette. The tissue digested was transferred to a 5-ml centrifugation tube on ice, and the remaining small pieces of ventral horn gray matter were further digested in trypsin-EDTA (16 min). The total cell suspension was then centrifuged at different speeds for cell sorting.
Sorting of Cell Suspensions
To isolate a spinal motor neuron-enriched fraction, tissue digests were centrifuged (Beckman GPR model centrifuge) at 200 rpm (20 gav) for 5 min (4C). The supernatant was collected and then centrifuged at 400 rpm (50 gav), 800 rpm (160 gav), and then 2500 rpm (1400 gav). After each spin (for 5 min), the pellet was resuspended in 100 μl PBS, pH 7.4, fixed with 1 ml of 4% paraformaldehyde (4C for 1 hr) for cell characterization using immunocytochemistry, TUNEL, or cresyl violet staining.
Characterization of Sorted Cell Suspensions
The cells were repelleted after fixation and each pellet was resuspended with 250 μl PBS. An aliquot of cell suspension (50 μl) was applied to a gelatin-coated slide and a coverslip (24 mm × 30 mm) was gently overlaid to form a monolayer of cells. The slides were then air-dried. Air-dried slides were rinsed (1 hr) in PBS to separate the slides from the cover-slips. The cells did not attach to the coverslips because they were not coated with adhesive; instead, the cells were attached to the gelatin-coated slides. The cells were permeabilized (30 min) in 1% Triton X-100 and then treated (30 min) with 1% bovine serum albumin (BSA). The cells were probed with antibodies to neuronal nucleus protein (NeuN, diluted 1:20), a neuron-specific marker (Chemicon International; Temecula, CA), choline acetyltransferase (ChAT, diluted 1:5), a marker for motor neurons in rat spinal cord enlargements (Roche Molecular Biochemicals; Indianapolis, IN), glial fibrillary acidic protein (GFAP, diluted 1:20), an astroglial marker (DAKO; Glostrup, Denmark), CD11b/c IgG2a (OX-42, diluted 1:20), a microglial/macrophage marker (Harlan Sera-Lab; Sussex, UK). Diluted primary antibodies were applied to the slides and the slides were incubated (24 hr at room temperature) in a humidified box. After primary antibody incubation, the slides were rinsed in PBS. Alexa-conjugated anti-mouse IgG (diluted 1:100; Molecular Probes, Eugene, OR) was used to visualize NeuN, ChAT, and OX-42, and Cascade blue-conjugated anti-rabbit IgG (diluted 1:100; Molecular Probes) was used to visualize GFAP. The slides were incubated (4 hr at RT) in a humidified dark box. The slides were washed and coverslipped with propidium iodide/antifade (Ventana; Tucson, Arizona). The slides were observed and photographed under a Zeiss fluorescence microscope. The preparation of cell suspensions from rats used for retrograde tracing of motor neurons was identical to that described above. The slides were coverslipped with or without propidium iodide/antifade and were observed under the same fluorescence microscope but with UV emission.
Counting of Different Cell Types in Cell Suspensions
To identify the cell fraction that was enriched in motor neurons, neurons marked with cell-specific antibodies were counted. The total number of neurons was estimated by comparing the number of NeuN-positive cells to the total number of cells identified by propidium iodide staining plus NeuN staining. To determine the proportion of spinal motor neurons in the cell suspensions, the percentage of ChAT-positive cells relative to NeuN-positive cells was calculated. To determine the proportion of spinal motor neurons issuing sciatic nerve axons, the fraction of DAPI-positive cells relative to the total number of NeuN-positive cells was also calculated. The numbers of labeled cells from six different microscopic fields (×400) were averaged from each case and then a total mean was derived from the preparations from three different cases.
Immunoblotting
Immunoblotting was used to identify ChAT immunoreactivity in lysates of motor neuron cell suspensions to additionally verify the presence of a motor neuron phenotype in this fraction. The cells were pelleted, washed, and lysed in buffer. Samples (20 μg of total protein) were fractionated by SDS-PAGE. Proteins were electroeluted onto nitrocellulose sheets. Blots were washed with 50 mM Tris-buffered saline (TBS) and blocked in 2.5% nonfat milk in 50 mM TBS/0.1% Tween-20. ChAT immunoreactivity was detected with a monoclonal antibody (Incstar, Stillwell, MN) used at a concentration of 1 μg IgG/ml. Immunoreactivity was visualized with enhanced chemiluminesence.
In Vitro Exposure of Motor Neurons to ROS
We tested the hypothesis that adult motor neurons rapidly accumulate genomic DNA lesions in response to oxidative stress. Motor neuron-enriched cell suspensions were exposed to H2O2, NO donors, H2O2 + NO donor, and ONOO−. Two different NO donors were used: sodium nitroprusside (SNP; Sigma, St Louis, MO) and N-(2-amino-ethyl)-N- (2-hydroxyl-nitrosohydrazino) -1,2-ethylenediamine (spermine-NONOate; OXIS International, Portland, OR). Spermine-NONOate was used because this agent can maintain long exposure to steady-state generation of NO (Hrabie et al. 1993). Motor neurons were also exposed directly to ONOO− (Alexis; San Diego, CA). ONOO− is a potent and relatively long-lived ROS formed by a reaction between O2 − and NO (Beckman et al. 1993).
Motor neuron cell suspensions (the 400 rpm preparation) were prepared from naïve rats. Motor neurons were exposed to SNP at concentrations of 10, 100, and 300 μM for different durations ranging from 15 min to 4 hr. Identical cell suspensions were exposed to spermine-NONOate at concentrations of 10 and 100 μM for 30 min, 1 hr, and 2 hr. Alternatively, motor neurons were treated with ONOO− at concentrations of 10 and 100 μM for 15 min, 30 min, and 1 hr. These exposures were done in medium containing 90% Neurobasal-A (Gibco), 5% horse serum, 5% fetal bovine serum (both sera were heat-inactivated) and 1 × glutamine (Gibco) in a tissue culture incubator (containing 5% CO2 and 95% air, 37C) for the different times. For controls, samples of the same cell suspensions were incubated in medium for the same time in the absence of SNP, with spermine tetrahydrochloride/sodium nitrite (NO2 −) or with decomposed ONOO− in alkaline solution. After exposure, the treatment groups were collected in 5-ml centrifuge tubes and repelleted at 4C for 5 min. Each pellet was resuspended and subjected to the comet assay.
Comet Assay
Preparation of Cell Microgels on Slides. To detect DNA damage in individual cells, motor neuron cell suspensions that were exposed to H2O2, NO donor, H2O2/SNP, and ONOO− were analyzed by the comet assay. In addition, to identify DNA damage in motor neurons undergoing apoptosis in vivo, the comet assay was used on motor neuron cell suspensions prepared from rats with sciatic nerve avulsions. The 400 rpm cell preparations from ipsilateral or contralateral sides of ventral horns of lumbar enlargements of animals with unilateral sciatic nerve avulsions were subjected directly to comet assay immediately after they were sorted and repelleted. All the procedures for comet assay were done under low light to minimize spontaneous DNA damage.
Our method for the comet assay on motor neurons is based mainly on the original protocol for lymphocytes (Singh et al. 1988). Many details were modified in the procedure for our application. The cell microgels were prepared as layers. The first layer of gel was made by applying 200 μl of regular melting point agarose (0.7%) onto superfrosted glass microscope slides (3″ × 1″, thickness 1 mm) and a coverslip was laid gently on the agarose. The agarose was allowed to solidify at 4C and the coverslip was removed. Low melting-point agarose was prepared in 100 mM PBS and kept at 37C. Samples of the motor neuron-enriched cell suspension were mixed with the low melting-point agarose and 50 μl of a mixture of cell suspension (containing ∼4.4 × 104 motor neurons) and low melting-point agarose was applied to the first gel layer. The slides were then coverslipped and placed at 4C for solidification of the cell suspension-agarose mixture. After the second layer solidified, the coverslips were removed and 100 μl of low melting-point agarose was added on top of the cell layer. The gels were re-coverslipped and the slides were placed on ice for gel solidification.
For preparing microgels, we compared two brands of low melting-point agarose (at the same concentrations) from different companies: Gibco BRL LMP agarose (cat. no. 15517–022) and FMC BioProducts SeaPlaque GTG agarose (cat. no. 50110). We found that the FMC agarose was the best for our purpose.
Lysis of Cells, DNA Unwinding, Gel Electrophoresis, and DNA Staining. Coverslips were removed from the cell microgels and the slides were covered with 1.5 ml of lysis buffer at pH 10 (for alkaline conditions) or pH 8.6 (for neutralized conditions) containing 2.5 M NaCl, 100 mM EDTA, 1% sodium lauryl sarcosine, 10 mM Tris, and Triton X-100 (final concentration 1%, freshly added immediately before use). The cell microgels were lysed for 30 min (at RT). After draining, microgels were treated with DNA-unwinding solution (300 mM NaOH, 1 mM EDTA, generally at pH 12 unless otherwise stated) for 30 min at RT. In some experiments, the effects of DNA unwinding solution/electrophoresis buffer pH on comet patterns were studied. Three different pH conditions were used: pH ≥13, pH 12, and pH 7.4. Loss of a purine or pyrimidine base from the DNA sugar-phosphate backbone facilitates an alkali-catalyzed β-elimination of the 3′-phosphate (Kohn 1991). At pH ≥13, alkali-labile sites are converted to SSB (Kohn 1991). A pH of 12 is reported to be appropriate for SSB detection because hydrogen bonds destabilize at this pH and double-stranded DNA separates into individual single strands, rendering shorter single-stranded DNA (resulting from SSBs) more easily eluted from the nucleus (Tice et al. 2000). Neutral conditions are used to detect double-strand breaks because under neutral pH conditions DNA remains double-stranded and regions containing DSBs will allow the DNA to migrate more readily in an electrophoretic field (Singh et al. 1988; Kohn 1991). The microgels were then placed directly into a horizontal gel electrophoresis chamber filled with DNA-unwinding solution. Gels were run with constant current (300 mA at RT) for usually 20 min. After electrophoresis, the microgels were neutralized with 50 mM Tris-HCl (pH 7.5) for 15 min (twice). DNA was visualized with ethidium bromide staining (20 μg/ml, 20 min at RT), after which the microgels were washed and coverslipped. The evaluation and image acquisition were performed using a Zeiss fluorescence microscope.
Counting Comets in Microgels
The number of cells with comets were counted in microgels prepared from motor neurons exposed to ROS in vitro and from motor neurons isolated from in vivo axotomy experiments. For cell preparations exposed to H2O2, NO donor, H2O2/SNP or ONOO−, cells incubated for the same time in medium without oxidant were used as controls. Three to five separate experiments from different animals were done for each type of in vitro oxidant exposure experiment. For the unilateral sciatic nerve avulsion experiments, comet assays were performed on two to four rats for each recovery time (5, 7, 10, 14, or 28 days). The contralateral (unlesioned) side of the spinal cord from rats with sciatic nerve avulsions was used as the control for each time point. The number of comets and large intact cell nuclei regarded as motor neurons stained by ethidium bromide were counted in six microscopic views at ×200 from microgels of treated cells and from sciatic nerve avulsion animals. The percentages of comets relative to the total number of cells (total number of comets and total number of intact cell nuclei) were determined and group means were calculated. The data were analyzed using a Student's t-test.
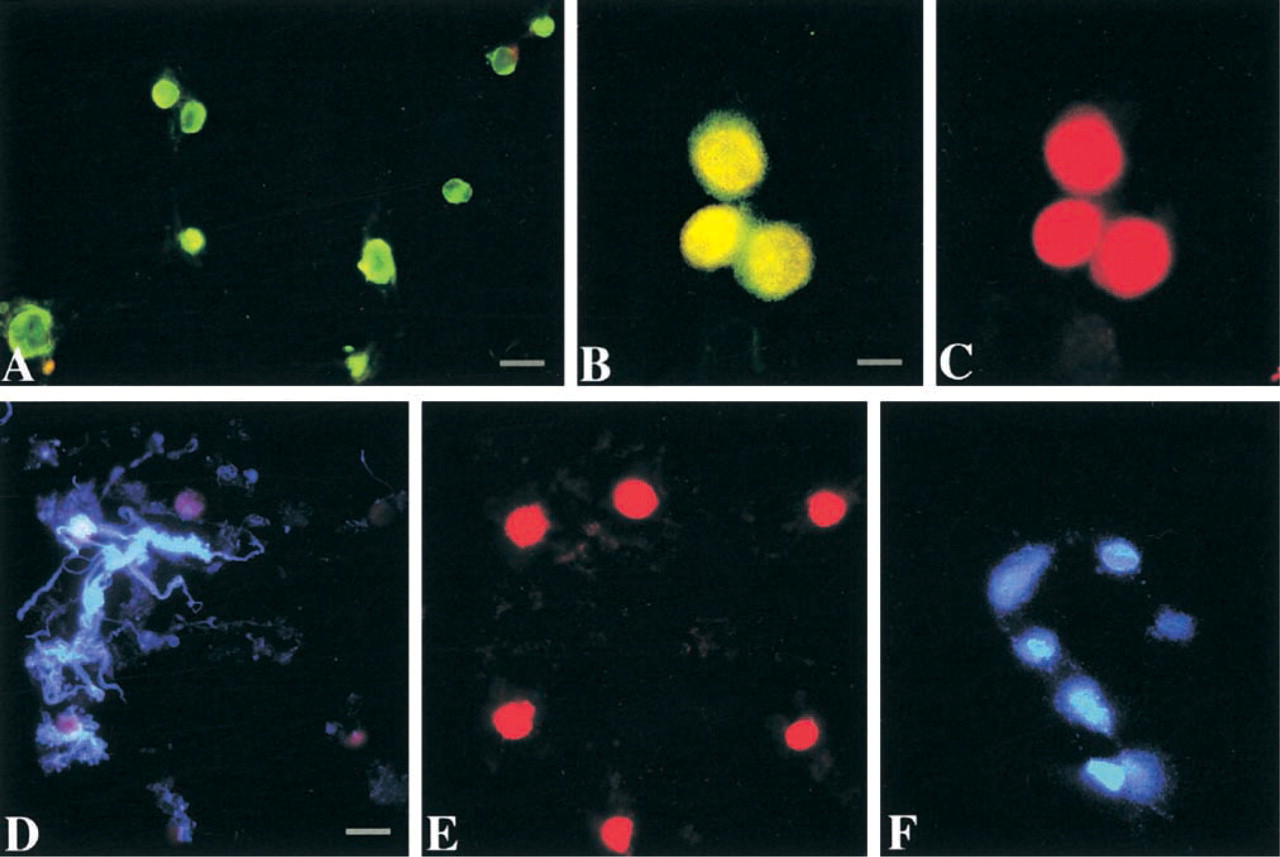
Characterization of the motor neuron-enriched cell suspension isolated from adult rat spinal cord ventral horn. (
TUNEL
Motor neuron cell suspensions exposed to NONOate (10 μM or 100 μM) or corresponding vehicle (10 μM or 100 μM spermine/NO2 −) were analyzed with TUNEL. Cells were fixed in 4% paraformaldehyde overnight. They were pelleted and washed with PBS (pH 7.4), and then mounted on gelatin-coated slides by coverslipping to form a cell monolayer and allowed to air-dry. The slides were rinsed in PBS for 1 hr to separate the coverslip from the slides. A modified enhanced TUNEL procedure was used. The cells were rinsed in 1% Triton X-100 for 1 hr and washed with PBS before they were rinsed with terminal deoxynucleotidyl transferase (TdT) buffer (containing 30 mM Tris, pH 7.2, 140 mM sodium cacodylate, 3 mM cobalt chloride) for 20 min. The TdT buffer was changed to fresh TdT buffer (0.5 ml/slide) containing TdT (0.02U/μl) and biotin-16-dUTP (50 μM) (both reagents were from Roche Molecular Biochemistry) and the slides were incubated at 37C for 2 hr. The reaction was terminated by incubating the slides in SSC (300 mM sodium chloride, 30 mM sodium citrate) for 15 min. The slides were then incubated in avidin–biotin–peroxidase complex for 2 hr at RT. After washing with PBS, the slides were stained with DAB/H2O2 for 20 min. Some of the slides were counterstained with cresyl violet and were observed and photographed.
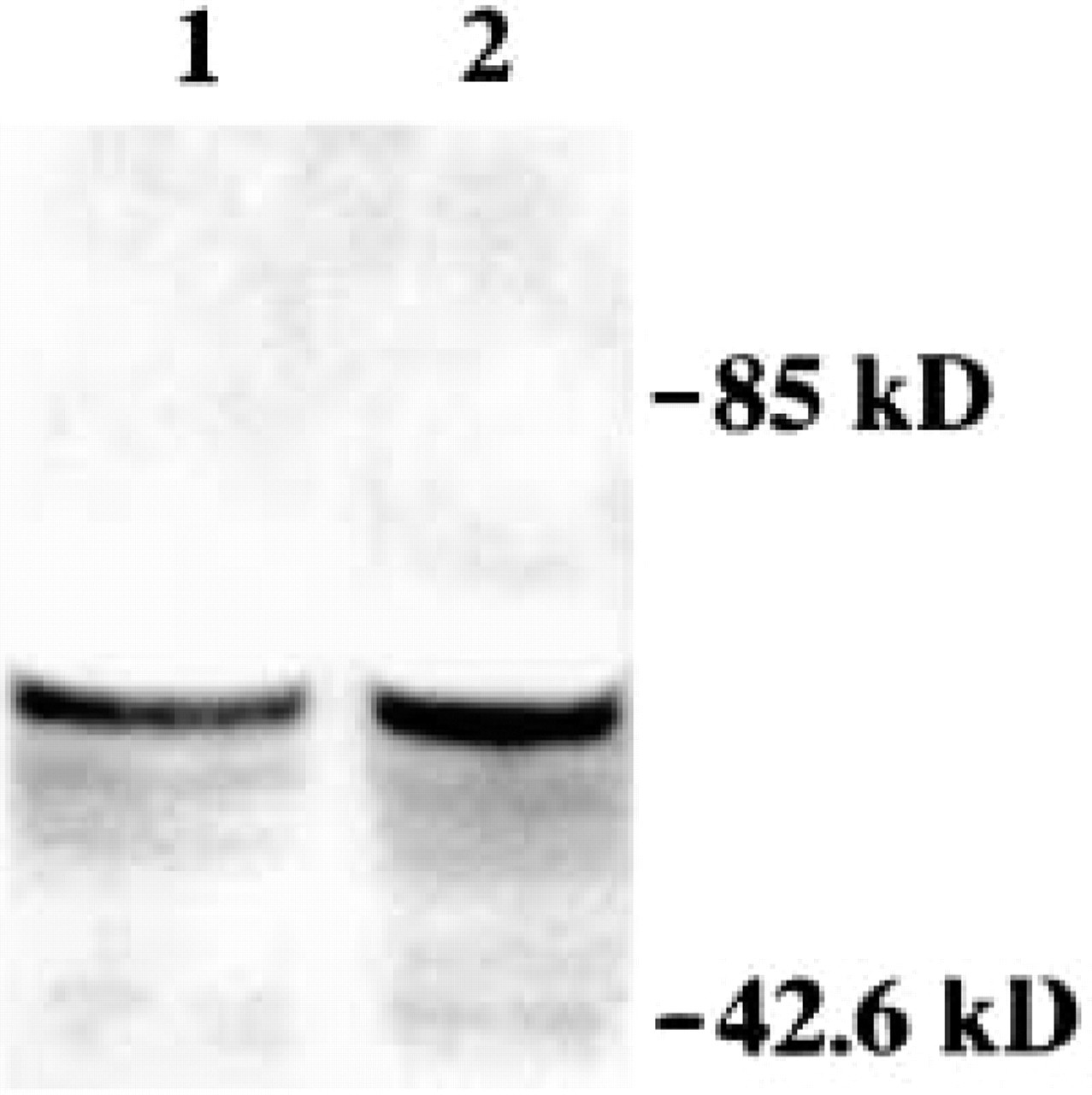
Immunoblot for ChAT in motor neuron cell suspensions. ChAT (detected as a major band at ∼67 kD) is enriched in lysates of cell suspensions, confirming the presence of a motor neuron phenotype. Molecular mass markers (in kD) are indicated at right. The levels of immunoreactivity are similar in motor neurons incubated for 30 min with 100 μM ONOO− (Lane 1) or vehicle (Lane 2), indicating no generalized degradation of cells.
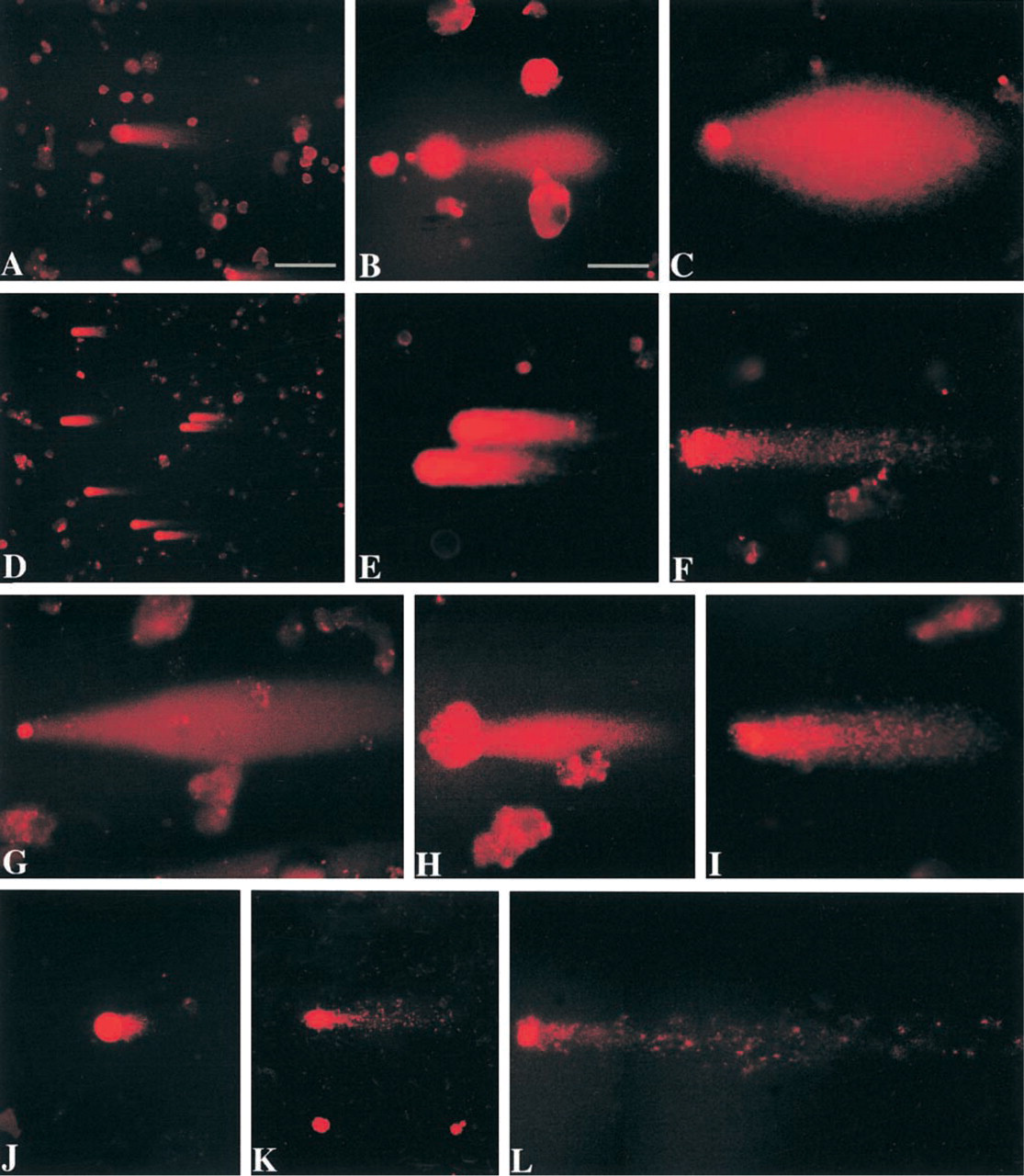
Alkaline (pH 12) comet assay on isolated motor neurons after exposure to ROS or axotomy. (
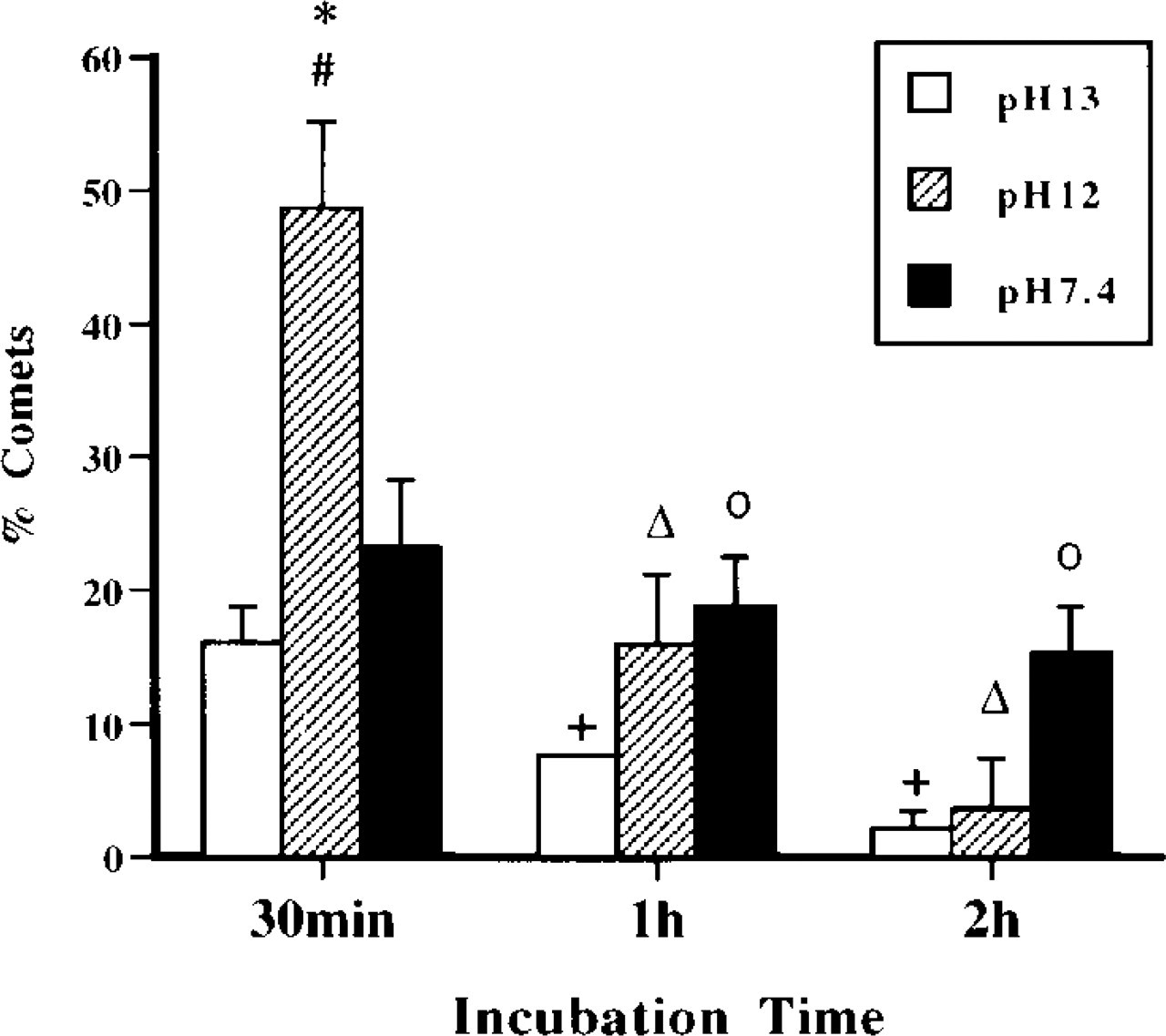
Histogram of percentage of comets detected using different pH conditions for the comet assay. Motor neurons were incubated in MEM alone for 30 min, 1 hr, or 2 hr. Alkali-labile sites/SSBs (at pH 13), SSBs (at pH 12), and DSBs (at pH 7.4) all exist in spontaneously degenerating motor neurons. Motor neurons with SSBs are significantly more numerous at 30 min compared to comet numbers at pH 13 (∗, p < 0.05) and pH 7.4 (#, p < 0.05). With increased incubation time, the number of comets visualized at pH 13 and pH 12 decreases, indicating a decrease in the formation of AP sites (+, p < 0.05 for pH 13 compared to earlier pH 13 time point) and SSBs (Δ, p < 0.05, pH 12 compared to 30-min pH 12 time point). In contrast, the number of comets detected at pH 7.4 remains comparatively stable, indicating that the level of DNA DSBs is relatively invariant. Therefore, the detection of AP sites and SSB is transient. °, p < 0.05, pH 13, compared to time matched pH 7.4 samples.
Double Labeling of Comet Assay Microgels with Immunocytochemistry
We evaluated the feasibility of characterizing motor neuron comets by immunocytochemistry. Motor neurons were examined for expression of survival motor neuron (SMN) protein and p53 in microgels after comet assay. Coverslips from microgels of motor neurons exposed to either H2O2 (for SMN) or NONOate (for p53) were removed after they were stained with ethidium bromide and observed for comets. Mouse monoclonal antibodies were used to localize SMN (Transduction Laboratories; Lexington, KY) and p53 (Santa Cruz; Santa Cruz, CA). Microgels were incubated with primary antibody dilutions (1:250 for SMN and 1:50 for p53) for 24 hr at RT. The slides were washed with PBS, then incubated (1:50–1:100 dilution) with Alexa-conjugated antimouse IgG for 2 hr at RT. Afterwards, the slides were washed and re-coverslipped for observation and photography.
Results
New approaches need to be developed to study mechanisms of degeneration of adult motor neurons. Specifically, sensitive quantitative assays for specific types of DNA lesions need to be identified to study the possible role of DNA damage in the upstream mechanisms of motor neuron apoptosis. We developed and characterized a novel short-term, motor neuron-enriched cell suspension system to evaluate the formation of DNA damage directly in individual adult spinal motor neurons.
Purification of a Motor Neuron-enriched Cell Suspension
Ventral horns of cervical/lumbar enlargements of adult rat spinal cord were microdissected and subjected to mild dissociation with trypsin-EDTA. The total cell suspension was fractionated by low-speed centrifugation. The different cellular fractions were examined for NeuN, ChAT, GFAP, and OX-42 immunofluorescence, and for DAPI fluorescence to identify α-motor neurons giving rise to sciatic nerve axons. One fraction was identified that is enriched in motor neurons. In the 400 rpm fraction of digests of ventral horn enlargements, neurons comprise ∼84% of the total cell number (NeuN-positive cells/total cells, i.e., NeuN-positive cells and cells stained with propidium iodide; Figure 1A). Of these neurons, ∼86% are motor neurons (ChAT-positive cells/NeuN-labeled cells; Figures 1B and 1C). Of these motor neurons, ∼72% are α-motor neurons giving rise to sciatic nerve axons (by DAPI sciatic nerve tracing; Figure 1F). Some motor neurons are entirely or partially surrounded by fragments of astroglial processes (Figures 1D and 1C), but GFAP-positive cells are rarely present in this fraction. OX-42-positive microglial cells are present only occasionally (not shown). Immunoblotting evaluation of fraction for ChAT confirmed the presence of motor neurons in this fraction (Figure 2).
Quantification of DNA damage in motor neuron comets induced by different ROS in vitro a
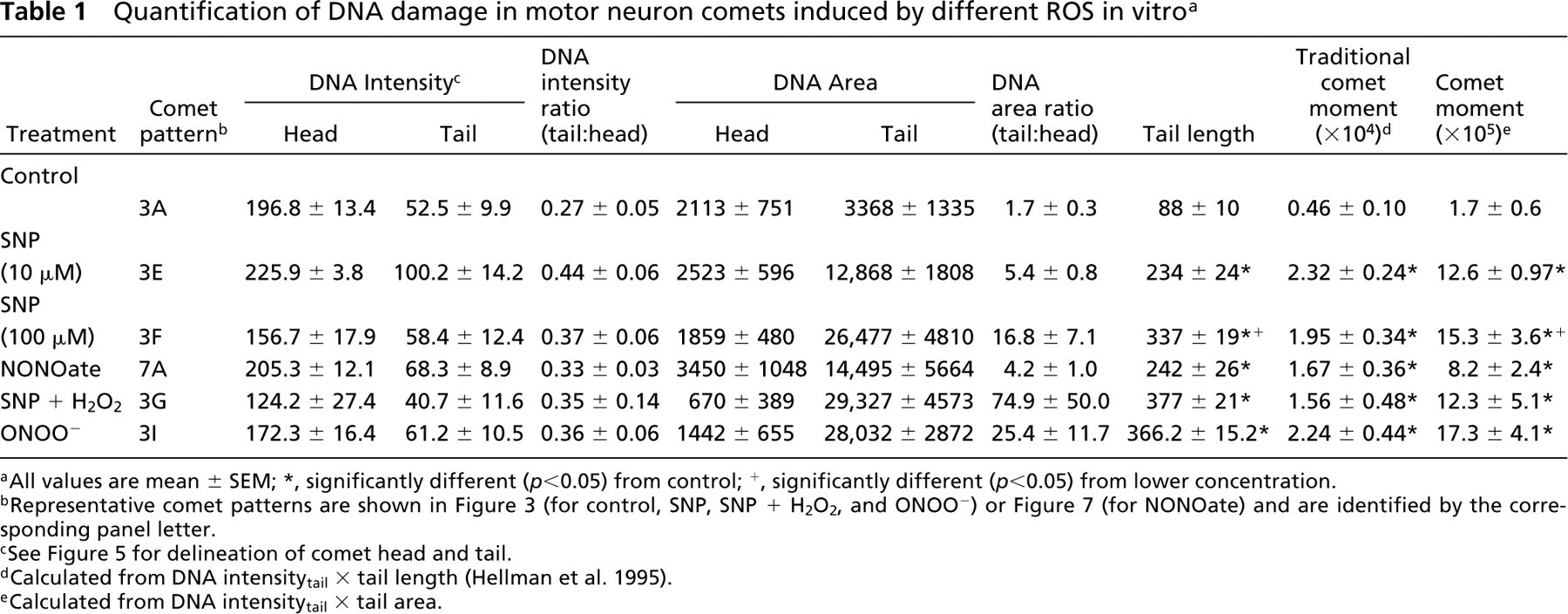
All values are mean ± SEM; ∗, significantly different (p < 0.05) from control; +, significantly different (p < 0.05) from lower concentration.
Representative comet patterns are shown in Figure 3 (for control, SNP, SNP + H2O2, and ONOO−) or Figure 7 (for NONOate) and are identified by the corresponding panel letter.
See Figure 5 for delineation of comet head and tail.
Calculated from DNA intensitytail × tail length (Hellman et al. 1995).
Calculated from DNA intensitytail × tail area.
DNA Damage Is Rapidly Induced and Accumulates in Motor Neurons Undergoing Oxidative Stress
Motor neuron cell suspensions were exposed to H2O2, NO donor, H2O2/SNP, or ONOO−. Control cells were exposed to vehicle and incubated in medium for the same time. Comet assays were done at pH 12 (at this pH hydrogen bonds in DNA destabilize, causing strand separation). Comets were observed with each treatment (Figure 3). Each treatment gave consistent results, with the major comet pattern generated from each exposure being highly reproducible (Figure 3). Figure 3 shows the most common patterns of comets after different treatments.
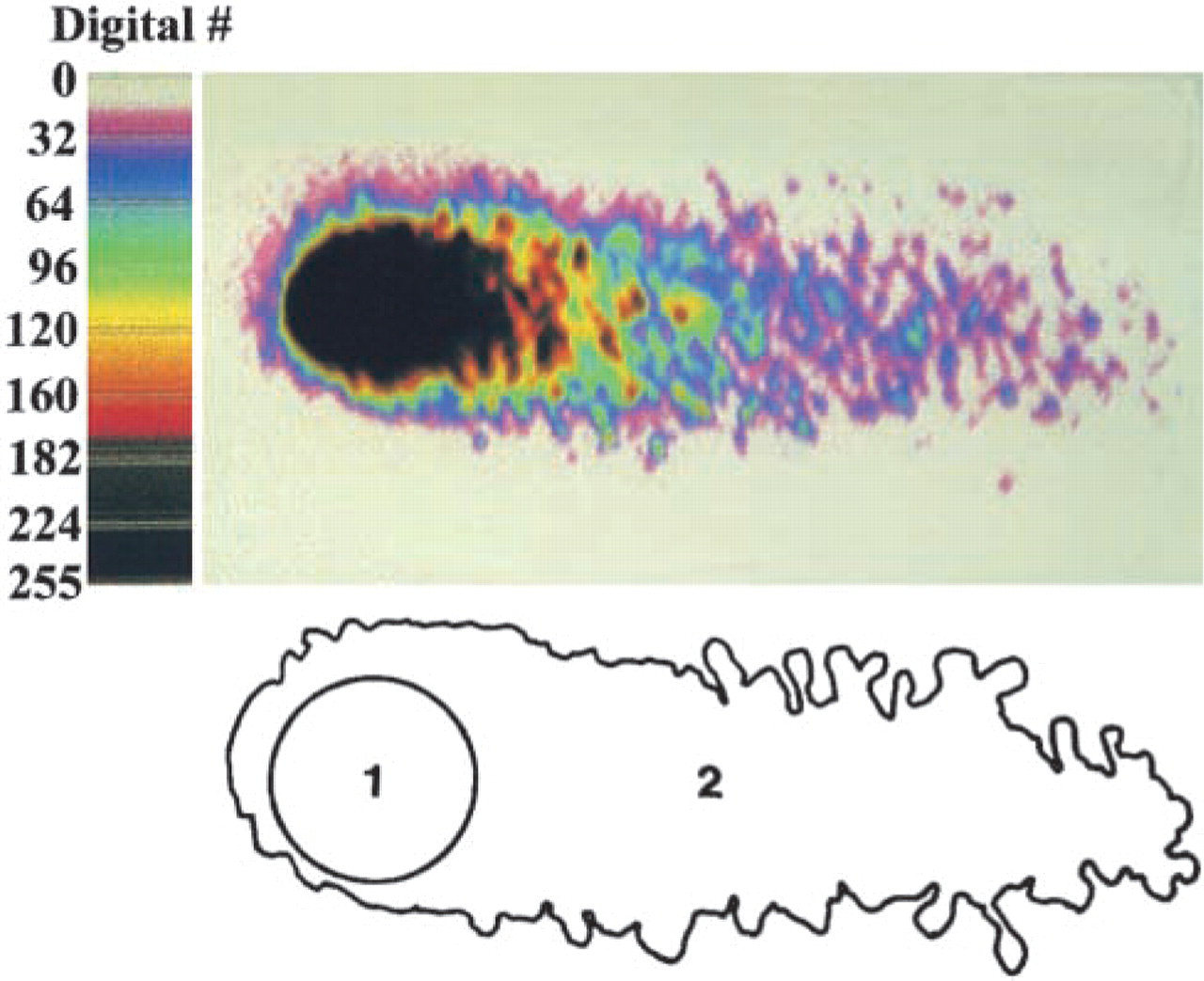
Measurement of DNA damage in motor neurons with the comet assay. Digital image (upper image) of a motor neuron comet (the tail and the head comprise the comet) found in micro-gels after single-cell gel electrophoresis. Ethidium bromide staining of DNA is in pseudocolor. Each color indicates a different DNA content, with black indicating highest DNA intensity (background color has the intensity of 0). Comet contour (lower image) delineating comet head (1) and tail (2).
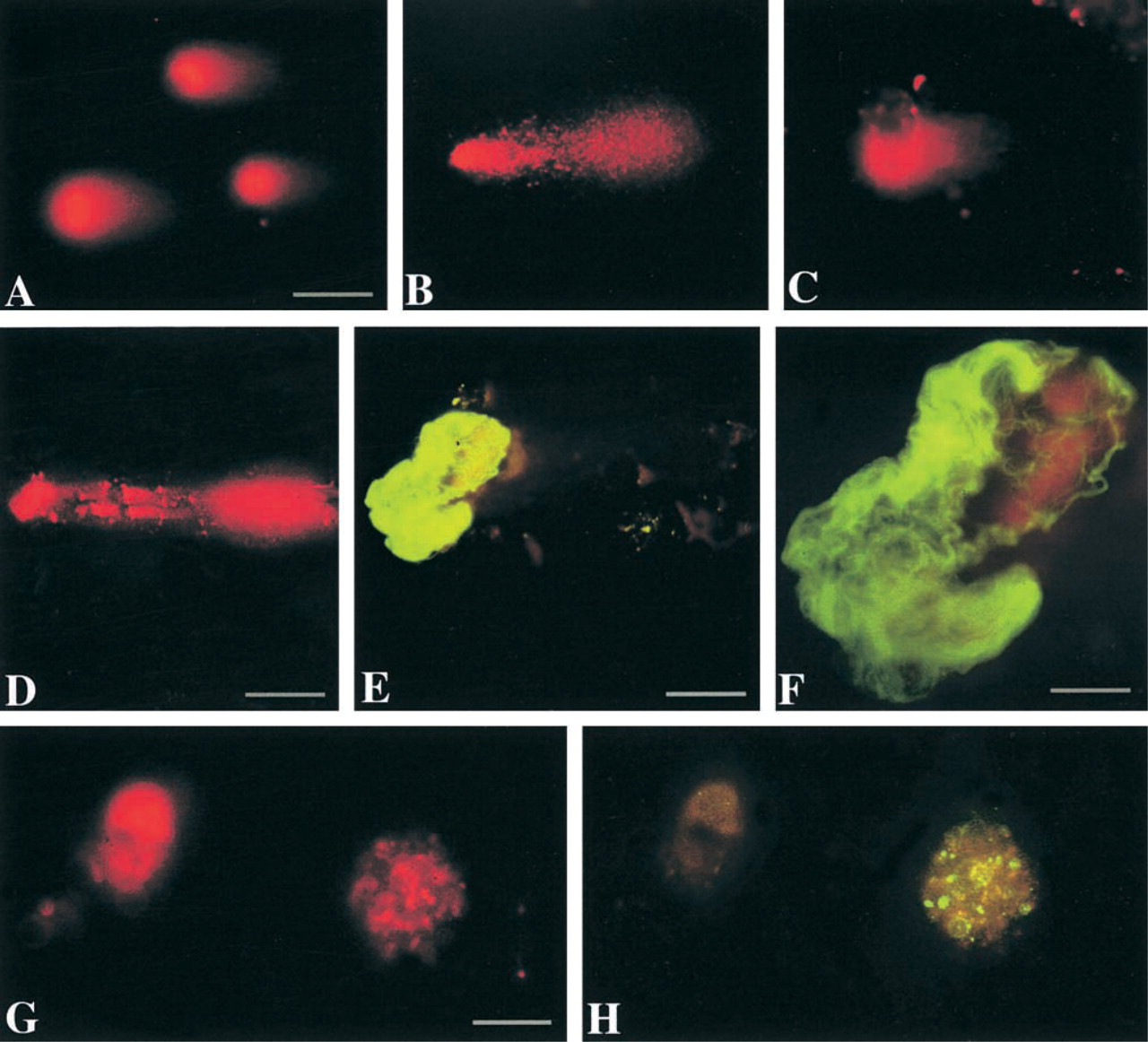
In microgels of control samples, the nuclei of many intact cells stained with ethidium bromide. Motor neurons with intact genomic DNA in gels stained with ethidium bromide have an evenly stained, smooth round nucleus without a tail (Figure 3A), indicating no DNA damage. In control motor neuron suspensions incubated in Neurobasal-A medium, the percentage of comets is ∼10% or less. Control comets have a large round head, densely labeled with ethidium bromide, and a short granular tail, composed of large scattered granules (Figure 3A). We have reported previously (Liu and Martin 2001) that viability of adult motor neurons in suspension is influenced by culture medium. Neurobasal medium is much better than MEM for maintaining adult motor neurons in vitro. Isolated mature motor neurons produce high levels of O2 − and spontaneously degenerate in MEM (Liu and Martin 2001). We used this system to evaluate the effects of DNA-unwinding solution pH on comet detection. At 30 min of incubation, many more motor neuron comets are observed at pH 12 compared to the number at pH 13 and 7.4 (Figure 4). In addition, the number of comets decreases over time (Figure 4). These results indicate that DNA SSBs are formed rapidly and accumulate transiently in dying motor neurons.
A comparison of control comets (Figure 3A) with comets induced by H2O2 (Figure 3B) and NO donors (Figures 3D-3H) shows the striking difference in the comet profiles and amount (Table 1) of DNA damage. With 1 mM H2O2 exposure in neurobasal-A medium, the comets have a round head, a tail with fine granules, and a short neck between head and tail (Figure 3B). With 1 mM H2O2 exposure in MEM, the comets have huge spindle-shaped tails with a small round head (Figure 3C), indicating severe DNA damage and elution from the cell. Exposure to NO donors generates consistent comet profiles depending on exposure concentration and duration. In general, with exposure to 10 μM SNP or NONOate for 15 min, 30 min, or even 1 hr, the comet head has a halo composed of large granules, with a short granular tail (Figures 3D and 3E). With 100 μM or higher doses, or with 10 μM for more than 1 hr of exposure, the head halo is less prominent but the tail length increases (Table 1), and it is still composed of scattered large granules (Figure 3F). Another comet pattern that shows severe DNA damage is found after motor neurons are exposed to 100 μM SNP with 1 mM H2O2 (Figure 3G). With these comets the head is small and the tail is long and wide; the degree of DNA damage is severe (Table 1). With increased exposure to 100 μM SNP with 1 mM H2O2, the comet head contains some clumped DNA, as shown by ethidium bromide staining (Figure 3H). We have found previously that the combination of SNP with 1 mM H2O2 induces severe protein nitration in motor neuron suspensions (Liu and Martin 2001), indicating the formation of ONOO− by the reaction of NO with O2 −. We therefore exposed motor neurons to ONOO− directly, and the majority of comets have granular heads and long granular tails (Figure 3I; Table 1). The DNA damage profiles of NO donors, NO donor plus H2O2, and ONOO− are similar.
Measurement of DNA Damage
The comet moment is regarded as one of the best indices of induced DNA damage in cells (Singh et al. 1988; Hellman et al. 1995). The extent of DNA damage was determined by measuring the displacement of DNA between the cell nucleus (i.e., the comet head) and the tail (Table 1). Cells with more DNA damage show an increased migration of DNA in the direction of electrophoresis (Östling and Johanson 1984; Singh et al. 1988). DNA migration length is believed to be directly related to fragment size, and it is believed migration is proportional to the level of SSBs and alkalilabile sites (Tice et al. 2000). DNA density and comet moments for individual motor neurons were measured using the fluorescence intensity of ethidium bromide-stained DNA (Figure 5). Randomly selected comets (∼10–30 comets in each treatment group) were captured as digital images using Inquiry software (Loats Associates; Westminster, MD). Comets were saved in TIFF format. Each comet was used to obtain several measurements by delineating the region of interest (Figure 5). DNA intensityhead, DNA intensitytail, areahead, and areatail were measured. Tail length was measured from the center of the head to the end of the tail. Relative DNA content is reflected by the average integrated intensity of fluorescence. The amount of DNA damage for each cell is derived from the calculation of the comet moment (Hellman et al. 1995; Petersen et al. 2000). We calculated the comet moment by two different methods. The traditional calculation is DNA intensitytail × tail length (Hellman et al. 1995). Because this equation does not take into consideration the broadness of the tail, we also calculated the comet moment from DNA intensitytail × areatail.
Comet assay analysis of different forms of DNA damage induced in motor neurons exposed to NO donor. (
Comet morphology at different pH conditions and comet assay combined with immunocytochemistry in microgels. (
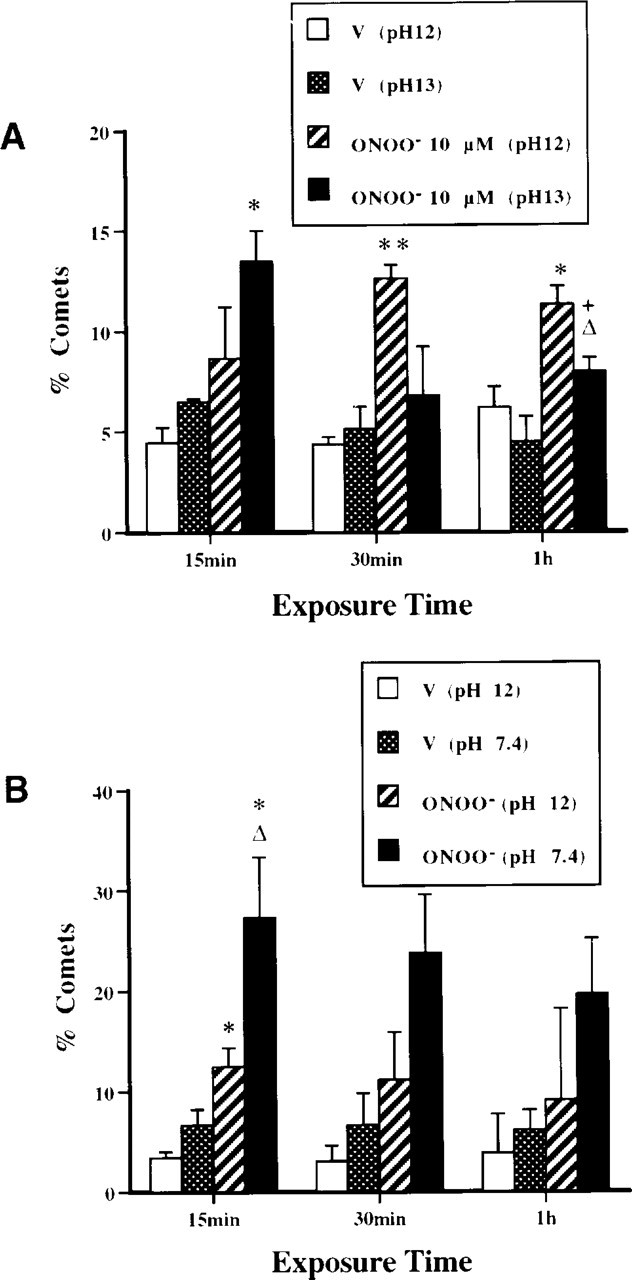
Comet assay analysis of different forms of DNA damage induced in motor neurons exposed to ONOO−. (
The comets in control motor neurons have low moments (∼1.7 × 105; Table 1). The comet moments in motor neurons exposed to ROS are significantly higher than those of controls (Table 1). These measurements show that NO, NO plus H2O2, and ONOO− induce DNA damage directly in motor neurons. In addition, the results show that the comet moment is a sensitive measure of DNA damage in motor neurons.
DNA Damage Occurs Early in Motor Neurons Undergoing Apoptosis In Vivo
We applied the comet assay to an in vivo model of motor neuron apoptosis in adult spinal cord. Sciatic nerve avulsion in adult rat causes apoptosis of lumbar motor neurons over 7–14 days (Martin et al. 1999). To identify whether DNA lesions occur in motor neurons early during the progression of apoptosis in vivo, the comet assay was used on motor neurons isolated from rats with sciatic nerve avulsions. With the same dissection and dissociation as mentioned above, the ipsilateral and contralateral motor neuron columns in the lumbar enlargement from sciatic nerve-avulsed animals were separately isolated and directly analyzed with the comet assay after cell sorting. We detected DNA damage in lumbar motor neurons injured by sciatic nerve avulsion. Motor neurons from the contralateral side did not show comets at any time. In contrast, as early as 5 days after sciatic nerve avulsion, the comet assay shows DNA damage, with comet profiles having a large head and a very short tail (Figure 3J). At 7 days after sciatic nerve avulsion, the comet head is looser and tail is longer (Figure 3K) than at 5 days. At 10 days after sciatic nerve avulsion, some of the comets have small heads and very long granular tails (Figure 3L), whereas others have a head with a big halo and a short granular tail (Liu and Martin 2001). Both are similar to the comet patterns observed after motor neurons are exposed to NO donors (Figures 3D–3F). At 14 and 28 days after sciatic nerve avulsion, no comets are observed in motor neurons from either ipsilateral or contralateral lumbar ventral horns.
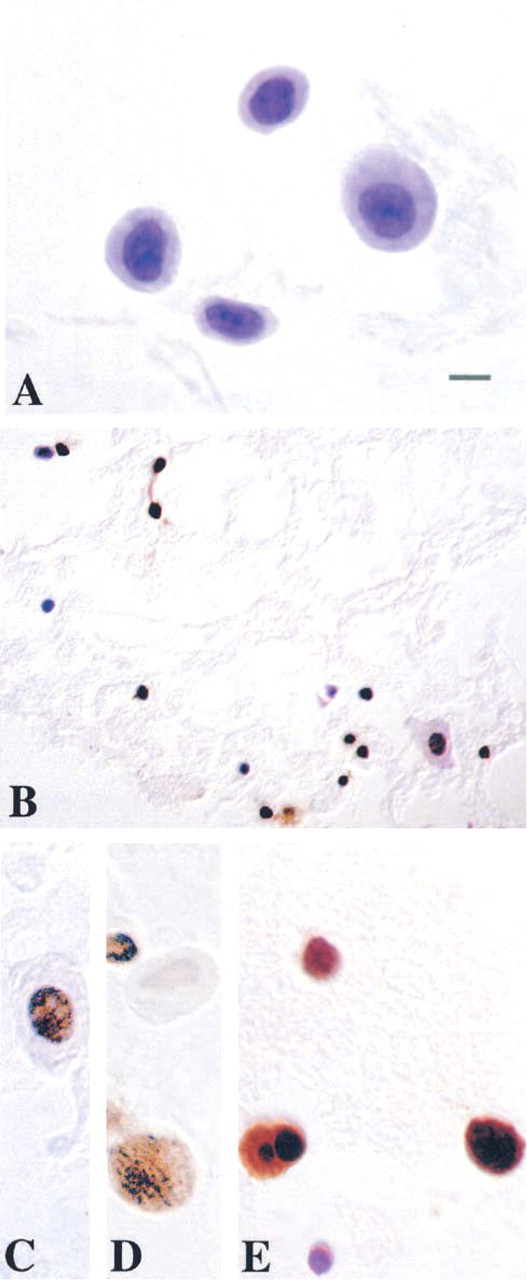
DNA fragmentation in motor neurons exposed to NO donor as analyzed by TUNEL Adult motor neurons were isolated, exposed in vitro to 10 μM NONOate or vehicle for 30 min, washed, and mounted on slides. (
Identification of Different Types of DNA Damage in Motor Neurons by Varying pH Conditions
The use of different pH conditions during electrophoresis is an approach to discriminate between DNA strand breaks and alkali-labile sites (Tice et al. 2000). This approach has been validated (Tice et al. 2000) but it has not been applied to neurons. At pH ≥13, alkali-labile sites are thought to be converted into SSBs, and DNA crosslinking diminishes migration of DNA strand breaks. At pH 13, we did not observe comets in motor neurons exposed to 10 mM H2O2 in neurobasal-A for several durations (30, 45, 60, and 90 min). At pH 12, many comets are observed after exposure of motor neurons to 10 mM H2O2 (Liu and Martin 2001). These results with neurons are consistent with results from non-neuronal cells showing that H2O2 mainly induces SSBs (SSBs:DSBs ∼1:2000) instead of DSB or alkali-labile sites (Singh et al. 1988; Horváthová et al. 1999). We conclude that comet assay conditions using pH 12 electrophoresis buffer are appropriate for detecting primarily DNA SSBs in motor neurons induced by H2O2.
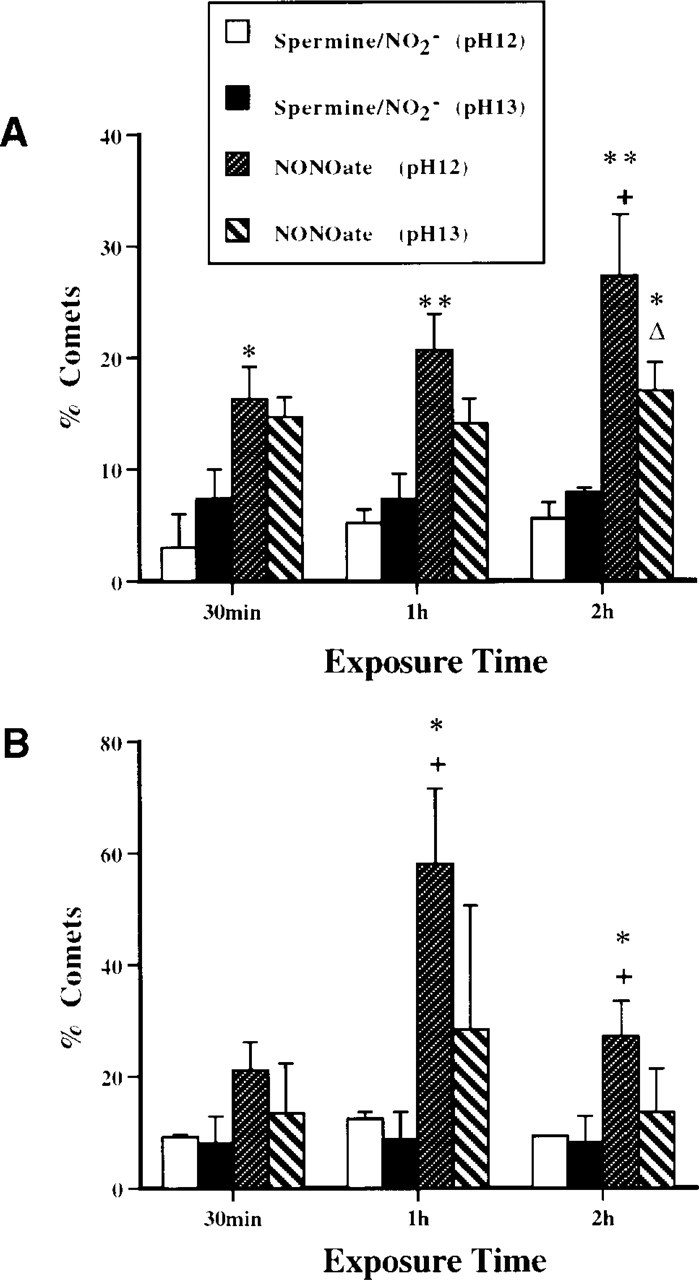
We evaluated the types of DNA damage (SSBs or alkali-labile sites) induced in motor neurons exposed to NO (Figure 6). At two different concentrations of NONOate (10 and 100 μM, exposure for 1 or 2 hr), the number of comets detected at pH 12 was generally greater than the number observed at pH 13. The comet morphology was different at pH 12 and, compared to pH 13 (Figures 7A and 7B). At pH 12, comets had prominent halos and short tails (Figure 7A), and comets at pH 13 had no halos and long granular tails (Figure 7B). At pH conditions of 12.6 or higher, alkali-labile sites are converted to SSBs and, therefore a pH ≥13 maximizes the detection of alkali-labile sites as SSBs (Kohn 1991). The presence of increased DNA migration from motor neurons at pH 13 indicates specifically the induction of alkali-labile sites, as well as SSBs, by NO donor. The formation of DNA SSBs is very dynamic. Exposure of motor neurons to 10 μM causes a progressive accumulation of DNA SSBs over a 2-hr period, while the level of alkali-labile sites is relatively invariant (Figure 6A). With exposure of motor neurons to 100 μM NONOate, a prominent peak in the number of comets detected at pH 12 is observed at 1 hr (Figure 6B). The DNA SSBs are formed within a 30-min time window. Therefore, motor neurons exposed to NO rapidly form DNA SSBs and alkali-labile sites.
The types of DNA damage were analyzed in motor neurons exposed to ONOO− (Figure 8). ONOO− induces rapid formation of alkali-labile sites, followed by an accumulation of SSBs while alkali-labile sites decline (Figure 8A). In addition, ONOO− induces DSBs (pH 7.4) very quickly in motor neurons (Figures 7C and 8B). These changes in motor neuron DNA were not the result of generalized cellular degradation because ChAT levels remained stable (Figure 2). Therefore, ONOO− is a potent DNA-damaging agent that concurrently induces alkali-labile sites, SSBs, and DSBs in motor neurons.
TUNEL Analysis of Isolated Adult Motor Neurons Exposed to NO Donor
TUNEL is a commonly used method for detecting DNA damage in cells. For comparison with the comet assay, we used the TUNEL method on motor neurons exposed to NO donor. Motor neuron cell suspensions were exposed to NONOate (10 μM or 100 μM) or corresponding vehicle (10 μM or 100 μM spermine/NO2 −). TUNEL confirmed that NO is toxic to motor neurons because many motor neurons were TUNEL-positive after exposure to NONOate (Figures 9B-9E) but not after exposure to spermine/NO2 − (Figure 9A). TUNEL staining ranged from light (Figures 9C and 9D) to very dark and aggregated (Figure 9E). However, the types of DNA damage could not be ascertained with the TUNEL method; yet, a major advantage of the TUNEL method over the comet assay is that the morphological progression of motor neuron apoptosis is better appreciated with the TUNEL method (Figure 9E).
Combination of Comet Assay with Immunocytochemistry
We evaluated whether comet assay and immunocy-tochemical techniques could be combined. This combination is desirable because information on the expression of specific proteins is helpful for understanding the mechanisms of DNA damage-induced motor neuron apoptosis (Martin et al. 2000). Survival motor neuron (SMN) protein, a motor neuron protein that functions in cell survival (Liu and Dreyfuss 1996), is detectable in microgels after the comet assay in the 400 rpm cell preparation (Figures 7D-7F), further confirming that these cells are motor neurons. Although cytoplasmic proteins are degraded by the lysis-procedure, SMN has a nuclear localization and appears fibrous under alkaline conditions (Figure 7F). p53 immunoreactivity is also detected in the cells after comet procedures. p53 is a known DNA-binding protein. Cells without tails showed p53 immunoreactivity. Some positive cells had faint labeling, with a nucleus that was small and shrunken with DNA condensed after the SSB stage (Figure 7G). Other cells had nuclei with p53 granules, some of which co-localized with DNA granules (Figure 7G and 7H). This pattern was observed frequently in motor neurons exposed to NO donor.
Discussion
This work advances the study of motor neurons in three ways. It shows that mature spinal motor neurons can be isolated and used for in vitro models of neurotoxicity. This demonstration was extended by showing that adult motor neurons can also be isolated from in vivo models of motor neuron degeneration and evaluated for DNA damage using single-cell analysis. This study also shows for the first time that the comet assay is a useful method for measuring distinct DNA lesions in individual motor neurons. Using the comet assay with different pH conditions, we identified the coexistence of different types of early DNA damage in motor neurons. This approach may enable the study of motor neuron disease to move in new directions, particularly with regard to understanding mechanisms of DNA damage-induced apoptosis of motor neurons, which is relevant to ALS (Martin 2000).
Mature Motor Neurons Can Be Studied In Vitro
We developed a new approach to study mature motor neurons by creating a short-term, motor neuron-enriched cell suspension isolated from spinal cord ventral horn enlargements of adult rat. The motor neuron enrichment of this cell system was confirmed by immunophenotyping (e.g., ChAT, NeuN, and SMN), retrograde tracing, and immunoblotting. Electron microscopy has also been used to confirm the presence of motor neurons in this preparation (Liu and Martin 2001). This isolation technique for adult motor neurons can be applied successfully to paradigms of in vivo motor neuron injury (e.g., axotomy) to understand mechanisms of motor neuron degeneration using in vitro assays.
We took advantage of the structural features (i.e., their large size) of adult motor neurons and the large difference in size between spinal motor neurons and surrounding cells in the ventral horn to assist in their isolation by centrifugation. With strict microdissection and appropriate dissociation, our cell-sorting method may also be useful for isolating other populations of selectively vulnerable neurons, including cerebellar Purkinje cells, CA1 pyramidal neurons, and nigral dopaminergic neurons. This procedure could be highly applicable for Purkinje cells because these neurons are much larger than any adjacent cells, although the appropriate centrifugation speeds and times need to be identified for neuronal types other than spinal motor neurons.
This method is an important technical advancement in the field of motor neuron degeneration because very few methods are available to study motor neurons in vitro. Embryonic motor neuron cultures are a widely used in vitro system (Henderson et al. 1995), but these are immature neurons in a low-density culture. A recognized limitation of our model is the relatively short viability of adult motor neurons in suspension (∼40% survival at 24 hr after isolation; Liu and Martin 2001), but long-term viability is also a problem with embryonic motor neurons, such that these cells are usually studied for only 24–48 hr after plating. There are several advantages of our mature motor neuron system. First, the cells are adult neurons and, because neuronal maturity appears to influence cell death mechanisms (Portera-Cailliau et al. 1997), this system may be more relevant than embryonic cell models for understanding motor neuron degeneration in ALS. Second, injured motor neurons can be isolated from adult animals after in vivo spinal cord or peripheral nerve manipulations. Similar experimental manipulations cannot be done with embryonic systems. Third, our isolation technique yields a high density of motor neurons (i.e., large number of cells) that can be used not only for comet assay and immunocytochemical methods but also for biochemical assays (e.g., immunoblotting and Southern blotting). Fourth, this isolation approach can be extended to adult transgenic mouse models of motor neuron degeneration. Fifth, our in vitro cell system can be used as a high-throughput screening system for environmental, synthetic, or biological compounds for neurotoxic (genotoxic), neuroprotective, or survival activities on adult motor neurons.
Early DNA Damage in Motor Neurons Can Be Measured with the Comet Assay
Since Kohn discovered that single-strand DNA is eluted rapidly under the alkaline condition from cells onto filters, the alkaline elution method has been used to detect and measure low-level DNA damage in eukaryotic cells (Kohn et al. 1976). This principle guided the development of a new method for analyzing DNA damage at the level of individual cells using single-cell gel electrophoresis (Östling and Johanson 1984; Singh et al. 1988). The comet assay detects DNA SSBs at pH ∼12, DNA DSBs at neutralized conditions, DNA-DNA or DNA-protein crosslinking, alkali-labile sites that cause SSBs under alkali conditions (pH >12.6), and damage to purine and pyrimidine bases (AP sites). The comet assay is sensitive enough for detecting one break per 2 × 1010 Daltons of DNA in lymphocytes (Singh et al. 1988). However, very few studies have applied the comet assay to neurons (Lai and Singh 1995; Morris et al. 1999; Liu and Martin 2001). In our description here, we used two different models of motor neuron degeneration to evaluate the feasibility of the comet assay for detecting early low-level DNA damage.
For one approach, we isolated motor neurons using our new method and exposed these cells in vitro to different ROS, followed by analysis by the comet assay. Motor neuron-enriched cell suspensions were exposed to H2O2, NO donors, H2O2 + NO donor, and ONOO−. We tested the hypothesis that oxidative stress causes adult motor neurons to accumulate DNA damage. We used different pH conditions during electrophoresis to discriminate between DNA strand breaks and alkali-labile sites in motor neurons. Interestingly, we found that different ROS induce different DNA damage signatures in neurons, which has not been shown before. H2O2 induces primarily SSBs in motor neurons, consistent with previous reports using non-neuronal cells (Horváthová et al. 1999). NO donors induce alkali-labile (AP) sites and also SSBs, whereas ONOO− induces a combination of SSBs, DSBs, and AP sites.
In the other model we used an in vivo lesion that induces motor neuron apoptosis (Martin et al. 1999). After this lesion, motor neurons were isolated during the early progression of degeneration and analyzed by the comet assay. Motor neurons in the unlesioned side of spinal cord did not show comets at any time (Liu and Martin 2001). In the lesioned side, motor neurons at 5 days after sciatic nerve avulsion had DNA damage, as detected with the comet assay. At this time, TUNEL labeling is negative in motor neurons (Martin et al. 1999). The motor neuron comets have a large head and a very short tail at 5 days post lesion. DNA damage in motor neurons was also detected at 7 days after sciatic nerve avulsion. The comet heads are looser and tails are longer than at 5 days. At 7 days post lesion, the TUNEL method slightly detects DNA damage in the ipsilateral side of lumbar spinal cord after sciatic nerve avulsion (Martin et al. 1999). At 10 days post lesion, the comet assay reveals motor neurons with more advanced comets than at earlier time points, indicating accumulating DNA damage. Beyond 10 days, motor neuron comets were not detected in the 400 rpm fraction but were detected in higher-speed centrifugation fractions (Liu and Martin 2001), indicating that the cells are now lighter, and possibly hypoploid, due to the apoptotic process (Martin et al. 1999). An increase in DNA SSBs could be caused by an increased rate of DNA lesion formation or a reduction in DNA repair mechanisms (Lai and Singh 1995). The comets of motor neurons after avulsion and the comets of motor neurons exposed to NO donor in vitro are similar on the basis of comet morphology. Because avulsion-induced motor neuron death is apoptosis (Martin et al. 1999), comet patterns consisting of prominent DNA granules within the heads and tails are therefore signatures of apoptosis in neurons.
The comet assay has advantages over other more frequently used methods for detecting DNA damage (e.g., nick end-labeling methods such as TUNEL). It appears that assays for single-stranded DNA are more sensitive and specific than TUNEL for apoptosis. We have confirmed indirectly the sensitivity of comet assay detection of DNA SSBs as an early and sensitive marker for apoptosis. We found that DNA SSBs are visualized prominently in avulsed motor neurons at least 2 days before the detection of DNA fragmentation by TUNEL (Martin et al. 1999) and 5 days before round DNA-containing aggregates are detected in the nucleus (Liu and Martin 2001). The results indicate that the comet assay is more sensitive than the TUNEL method for showing DNA damage. Moreover, in motor neurons exposed to oxidative stress, DNA SSBs are detected prominently before morphological evidence for chromatin condensation and electrophoretic detection of internucleosomal DNA fragmentation (Liu and Martin 2001). In addition, the comet assay reveals the type of DNA damage (e.g., SSBs, DSBs, and AP sites), depending on the conditions. Another benefit of the comet assay is that comets can be classified into categories or stages of DNA damage, by morphology, by measuring tail length, and by digital image analysis of comets for DNA intensity and comet moment.
Previous studies using the comet assay have not combined this method with immunophenotyping of neuronal populations with DNA damage. We anticipated that some nuclear proteins would retain antigenicity in comet assay microgels. This suspicion was confirmed. Motor neurons contained SMN within the nucleus. In cell cultures (Liu and Dreyfuss 1996) and in rat spinal cord (Pagliardini et al. 2000), SMN has been localized in nuclear structures called gems that are associated with coiled bodies. SMN functions in the processing of pre-mRNA (Liu and Dreyfuss 1996). We found a nuclear localization in rat motor neurons as well and also report the novel observation that, after alkaline denaturation, SMN possesses an interesting fibrous structure. We also found that p53 is induced in motor neurons exposed to NO donor (Figure 7G) and that some of these cells are apoptotic based on morphology (Figure 7H) and TUNEL (Figure 9E). We also found that NO donors induce DNA SSBs and AP sites in motor neurons (Figure 6). Because DNA SSBs are potent activators of p53 (Jayaraman and Prives 1995; Levine 1997), early formation and accumulation of oxidative stress-induced DNA-SSB in adult motor neurons could be a primary signal for motor neuron apoptosis. However, cells expressing p53 generally did not display comets at pH 12 (as did cells expressing SMN), although nuclear morphology indicated apoptosis in many cells. It is possible that the DNA damage in these motor neurons advanced beyond SSBs, suggesting that p53 activation occurs later than SSBs, and therefore SSB could be an upstream signaling mechanism for motor neuron cell death as in non-neuronal cells.
Footnotes
Acknowledgments
Supported by grants from the US Public Health Service, the National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS34100), and National Institute on Aging (AG16282), and the Department of Defense, US Army Medical Research and Materiel Command (DAMD17-99-1-9553).