
Abstract
The peptide nucleic acids (PNAs) constitute a remarkable new class of synthetic nucleic acid analogues, based on their peptide-like backbone. This structure gives to PNAs the capacity to hybridize with high affinity and specificity to complementary RNA and DNA sequences and a great resistance to nucleases and proteinases. Originally conceived as ligands for the study of double-stranded DNA, the unique physicochemical properties of PNAs have led to the development of a large variety of research and diagnostic assays, including antigene and antisense therapy, genome mapping, and mutation detection. Over the past few years, PNAs have been shown to be powerful tools in cytogenetics for the rapid in situ identification of human chromosomes and the detection of aneuploidies. Recent studies have reported the successful use of chromosome-specific PNA probes on human lymphocytes, amniocytes, and spermatozoa, as well as on isolated oocytes and blastomeres. Multicolor PNA protocols have been described for the identification of several human chromosomes, indicating that PNAs could become a powerful complement to FISH for in situ chromosomal investigation.
B
Recently, this new type of oligomer has been introduced in cytogenetics. The properties of PNAs have allowed the development of fast, simple, and robust in situ assays, and the efficiency of PNA probes has been demonstrated on various types of cells.
Here we provide an overview of PNA properties and the techniques exploiting PNA technology in molecular genetics and cytogenetics.
PNA Chemistry and Properties
PNAs are synthetic DNA analogues in which the phosphodiester backbone is replaced by repetitive units of N-(2-aminoethyl) glycine to which the purine and pyrimidine bases are attached via a methyl carbonyl linker (Figure 1). This unique chemical makeup provides PNA with unique hybridization characteristics. Unlike DNA and RNA, the PNA backbone is not charged. Consequently, there is no electrostatic repulsion when PNA hybridizes to its target nucleic acid sequence, giving a higher stability to the PNA-DNA or PNA-RNA duplexes than the natural homo- or heteroduplexes. This greater stability results in higher thermal melting temperature (Tm) values than are observed for DNA-DNA or DNA-RNA duplexes (Jensen et al. 1997). An additional consequence of the polyamide backbone is that PNAs hybridize virtually independently of the salt concentration. Therefore, the Tm of PNA-DNA duplex is barely affected by low ionic strength. This significantly facilitates the hybridization with the PNAs. The unnatural backbone of PNAs also means that PNAs are particularly resistant to protease and nuclease degradation (Demidov et al. 1993). Because of this resistance to the enzyme degradation, the lifetime of PNAs is extended both in vivo and in vitro.
PNAs hybridize to cDNA or RNA in a sequence-dependent manner, according to the Watson-Crick hydrogen bonding scheme. In contrast to DNA, PNA can bind in either parallel or anti-parallel fashion and can hybridize with either single-stranded or double-stranded DNA. Homopyrimidine PNAs, as well as PNAs containing a high proportion of pyrimidine residues, bind to cDNA sequences to form highly stable (PNA)2-DNA triplex helixes displaying high Tm. In these triplexes, one PNA strand hybridizes to DNA through standard Watson-Crick base-pairing rules, while the other PNA strand binds to DNA through Hoogsteen hydrogen bonds. The resulting structure is called P-loops (Nielsen 2001). In addition, PNA-DNA hybridization is significantly more affected by base mismatches than DNA-DNA hybridization. A single mismatch in a mixed PNA-DNA 15-mer duplex decreases the Tm by up to 15C, whereas in the corresponding DNA-DNA complex, a single mismatch decreases the Tm by only 11C (Giesen et al. 1998). This high level of discrimination at the single-base level has indicated that short PNA probes could offer high specificity and has thus allowed the further development of several efficient PNA-based strategies for molecular investigations and diagnosis.
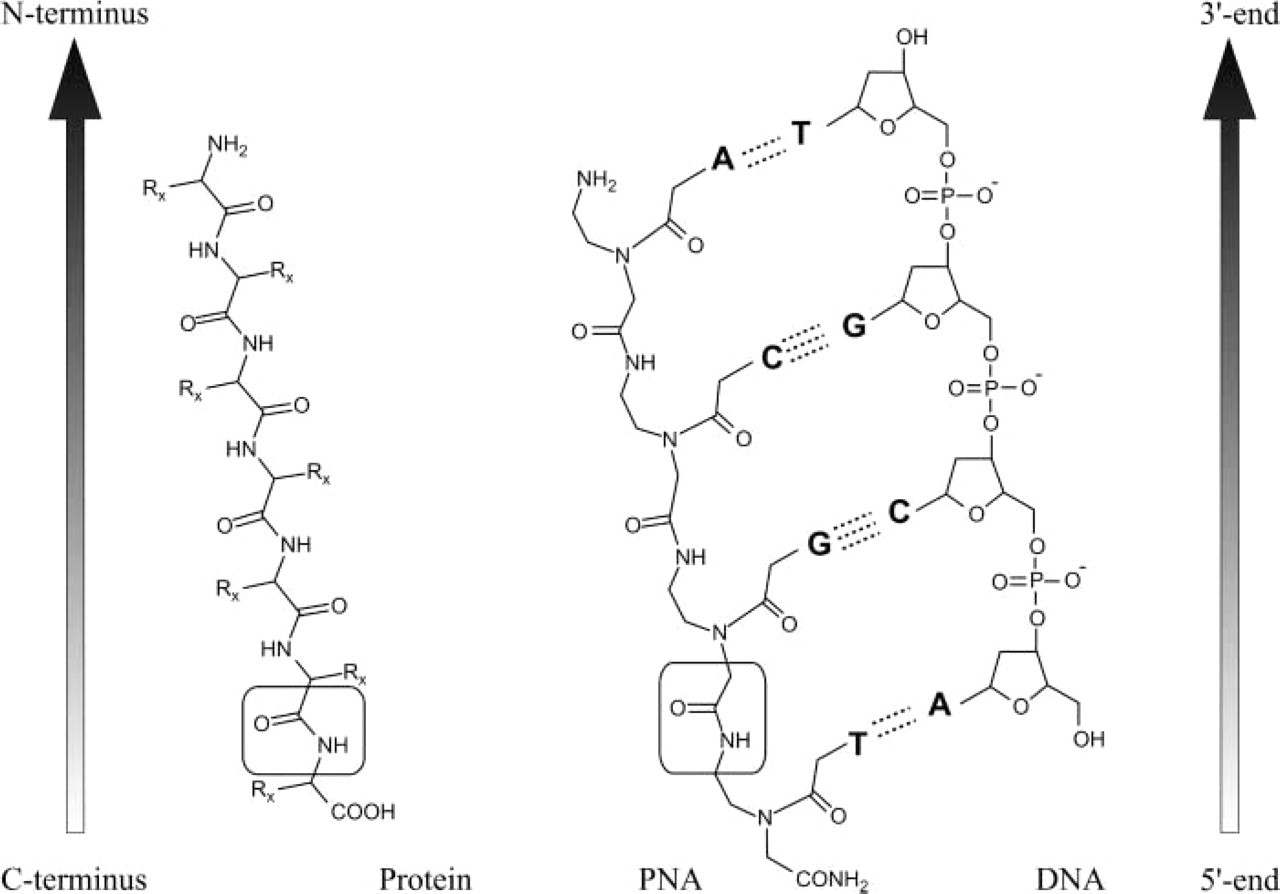
Chemical structures of PNA compared with DNA and protein. The backbone of PNA displays 2-amino-ethyl glycine linkages in place of the regular phosphodiester backbone of DNA, and the nucleotide bases are attached to this backbone at the amino nitrogens through methylene carbonyl linkages. The amide bond characteristic for both PNA and protein are shown in boxes. By convention, PNAs are depicted like peptides, with the N terminus at the left (or at the top) position and the C terminus at the right (or at the bottom) position. PNAs hybridize to cDNA or RNA sequences in a sequence-dependent manner, following the Watson-Crick hydrogen-bonding scheme. PNAs can bind to complementary nucleic acids in both parallel and anti-parallel orientation. However, the anti-parallel orientation illustrated in this figure is preferred. Watson-Crick hydrogen bonds are indicated by dotted lines.
Use of PNAs in Molecular Genetics
Since its introduction, an increasing number of applications of PNA technology have been described, confirming the high potential of peptide nucleic acids as efficient tools for molecular biology investigations.
PNAs as Antigene and Antisense Agents
PNA molecules were first used in antigene and anti-sense assays. Several in vitro studies demonstrated the ability of PNAs to inhibit both eukaryotic translation and transcription (Hanvey et al. 1992; Vickers et al. 1995; Boffa et al. 1996). PNA-mediated inhibition of gene transcription is mainly due to the formation of strand-invaded complexes or strand displacement in DNA targets. PNA targeted against the promoter region of a gene can form stable PNA-DNA complexes that restrict the DNA access of the polymerase, whereas PNA complexes located far from the promoter can block the polymerase progression and lead to the production of truncated RNA transcripts. Nielsen et al. (1994) have demonstrated that even an 8-mer PNA can efficiently block transcriptional elongation by (PNA)2-DNA triplex formation (Figure 2). Another way to alter gene transcription is based on the use of PNAs as competitors with endogenous cis-element(s) present in the target gene for trans-acting factors. This results in an attenuation of the authentic interactions of trans-factors with their cis-elements (Mischiati et al. 1999).
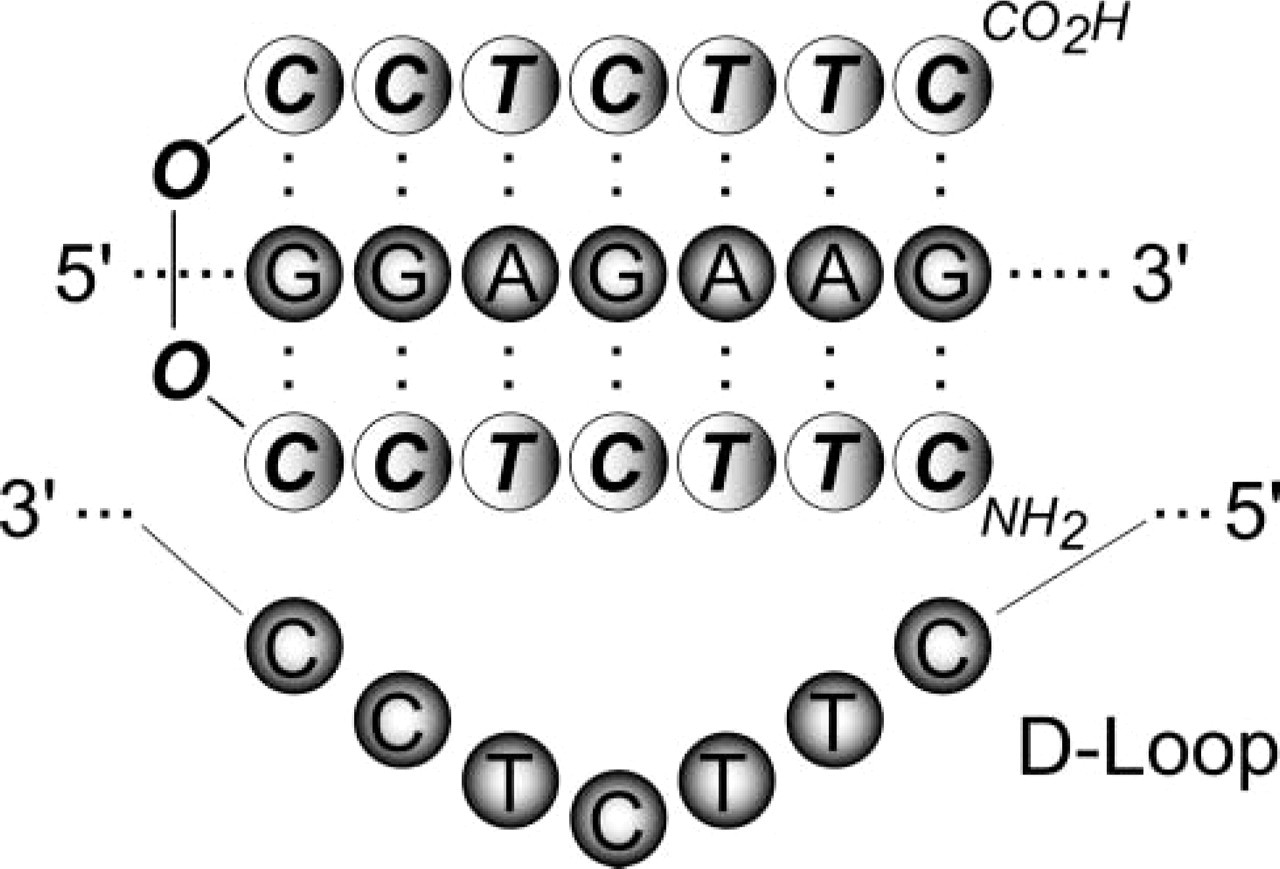
Triplex (PNA)2-DNA structure formed with cytosine-rich homopyrimidine PNAs binding to complementary homopurine DNA target. The PNA invades the DNA double-stranded helix, thus displacing a DNA strand into a “D-loop.” In this triplex structure, one PNA strand hybridizes to DNA through standard Watson-Crick base-pairing rules, while the other PNA strand binds to DNA through Hoogsteen hydrogen bonds. Base-pairing interactions are denoted by dots between the bases. O, linkers used to connect the two halves of the PNA oligomer.
PNAs are able to interact with mRNA independently of the RNA secondary structure. Studies on the mechanisms of antisense activity have demonstrated that PNA inhibits expression differently from anti-sense oligonucleotides acting through RNase-H-mediated degradation of the mRNA-oligonucleotide hybrid. Because PNAs are not substrates for RNase, their antisense effect acts through steric interference of either RNA processing, transport into cytoplasm, or translation, caused by binding to the mRNA (Knudsen and Nielsen 1996).
Despite the initial rapid success of PNA-based approaches in vitro, progress in the use of PNAs as tools for regulating gene expression was hampered by the slow cellular uptake of “naked” PNAs by living cells. Subsequent modifications of PNAs have led to significant improvements in the uptake of PNA in eukaryotic cells. The delivery into the cell can be speeded up by coupling PNA to DNA oligomers, to receptor ligands or, more efficiently, to peptides such as liposomes or cell-penetrating peptides that are rapidly internalized by mammalian cells (Pooga et al. 1998; Cutrona et al. 2000).
PNA-PCR Strategies
Naked PNAs have no direct interaction with DNA polymerase but can terminate the elongation of oligonucleotide primers by binding to the template or competing with the primers. Moreover, PNA-DNA chimeras can be recognized by the DNA polymerase and can therefore be used as primers for PCR reactions (Misra et al. 1998). PNA-DNA chimeras are covalently bonded hybrids in which a PNA oligomer is fused to a DNA oligomer to give rise to a new architecture exhibiting biological properties typical of DNA, such as the ability to act as substrate for enzymes.
The high-affinity binding of PNAs has also been used for detecting single base pair mutations by PCR. This strategy, termed PNA-directed PCR clamping, uses PNAs to inhibit the amplification of a specific target by direct competition of the PNA targeted against one of the PCR primer sites and the conventional PCR primer. This PNA-DNA complex formed at one of the primer sites effectively blocks the formation of the PCR product. The procedure is so powerful that it can be used to detect single base pair gene variants for mutation screening and gene isolation (Orum et al. 1993).
More recently, novel automated real-time PCR has been developed using PNAs. In this method, termed Q-PNA PCR, a generic quencher-labeled PNA (Q-PNA) is hybridized to the 5' tag sequence of a fluorescent dye-labeled DNA primer to quench the fluorescence of the primer. During PCR, the Q-PNA is displaced by incorporation of the primer into amplicons and the fluorescence of the dye label is liberated (Fiandaca et al. 2001).
Solid-phase Hybridization Techniques
The neutral backbone of PNAs significantly increases the rate of hybridization in assays in which either the target or the probe is immobilized. Therefore, PNAs can be used for sequence-specific capture of single-stranded nucleic acids, taking advantage of the tight complex formation at low ionic strength that destabilizes nucleic acid secondary structure. A system for capture of double-stranded DNA was also investigated using (PNA)2-DNA openers creating a large single-strand DNA loop to which a biotinylated oligonucleotide can hybridize. This complex allows the capture of the DNA via streptavidin beads (Bukanov et al. 1998).
The high-affinity binding of PNA oligomers might lead to faster and easier procedures in most standard hybridization techniques, such as Southern and Northern blotting (Nielsen and Egholm 1999). An alternative to Southern blotting analysis is the PNA pre-gel hybridization process, which significantly simplifies the procedure of Southern hybridization. Labeled PNAs are then used as probes, allowing hybridization to a denatured dsDNA sample at low ionic strength before loading on the gel. This is different from conventional Southern blotting, in which hybridization occurs after gel electrophoresis and membrane transfer. Here, the mixture is directly subjected to electrophoresis for separation of bound and unbound PNA probes. Because of their neutral charge, excess unbound PNA probes do not migrate in an electrical field. The PNA-DNA hybrids are then blotted onto a nylon membrane and detected using standard chemiluminescence techniques. The method is sensitive enough to detect a single mismatch in a DNA sample (Perry-O'Keefe et al. 1996).
Use of PNAs in Molecular Cytogenetics
During the past few years, the bulk of the interest in PNAs has focused on their exploitation as probes for ISH assays. Thanks to its high binding specificity, a single 15-mer PNA probe can substitute for a set of longer DNA probes. In addition, the neutral backbone of PNAs allows them to bind to DNA or RNA under conditions of low ionic strength, which discourages re-annealing of complementary genomic strands. This is particularly advantageous for in situ targeting of repeat sequences, for which both the length and the repetitive nature can favor re-naturation over hybridization with probes. Additional benefits of using PNAs are lower background signals and unlimited stability of the probe mixture (Williams et al. 2002). PNAs are compatible with a wide range of reporter molecules and fluorochromes, including fluoresceine, rhodamine, as well as cyanine and Alexa dyes available in a wide variety of colors.
The PNA-FISH technique was first used for quantitative telomere analysis. The study of telomere behavior has become a sensitive subject because of telomere involvement in the processes of cancer evolution and cellular senescence. The FISH technique has been successfully used for the in situ detection of telomeric repeat sequences in chromosomes of various species, using synthetic oligonucleotide probes, but the efficiency of these probes has not been sufficient to extend this procedure beyond qualitative analysis of repeat telomeric sequences. To monitor telomere length quantitatively, Lansdorp et al. (1996) used fluoresceine-labeled PNA probes. By comparing fluoresceine-labeled DNA, RNA, and PNA probes for the detection of telomeric repeat sequences on human metaphase chromosomes, they first showed that PNA probes yielded superior staining of telomeres. The PNA-FISH approach allowed the distinction of fluorescence of individual sister chromatid ends and the accurate estimate of individual and global telomere length in metaphase chromosomes of various cultured human hematopoietic cells. Subsequently, telomere PNA probes were used in several in situ studies of cancer and aging (Zijlman et al. 1997; Boei et al. 2000; Mathioudakis et al. 2000). The performance of the PNA method for in situ detection and sizing of telomeric repetitive sequences was compared with the primed in situ (PRINS) labeling technique. The two techniques were compared on mouse, hamster, and human cell lines, and the results were identical in terms of labeling efficiency and sensitivity (Serakinci and Koch 1999).
Further developments of PNA technology were focused on the improvement of the specificity of PNA probes and the in situ detection of numerical chromosome abnormalities. Chen et al. (1999) reported that PNA probes could discriminate between two centromeric DNA repeat sequences that differed by only a single base pair. Identical results were obtained with PRINS primers (Pellestor et al. 1995) and oligonucleotide probes (O'Keefe et al. 1996) but never with standard DNA probes. The identification of chromosomal variation and the analysis of polymorphisms could greatly benefit from the discriminative power of PNAs. The procedure of PNA synthesis allows consideration of the further production of allele-specific probes. This will constitute a marked improvement over the current labeling techniques.
Several chromosome-specific PNA probes have been designed and tested. Chen et al. (2000) defined short (15–18-mer) and specific PNA probes for alpha-satellite domains of nine chromosomes (chromosomes 1, 2, 7, 9, 11, 17, 18, X, and Y) and successfully used them on metaphase and interphase nuclei. To demonstrate the potential utility of PNA probes in clinical application, cultured and uncultured amniocyte preparations have also been analyzed, giving rates of hybridization efficiency of 90–97%. Taneja et al. (2001) tested other PNA probes for chromosomes 1, X, and Y, 18–22-mer in size and directly labeled with fluorochromes, on normal human lymphocytes and fibroblasts with abnormal chromosome contents. A fast and simple multicolor PNA protocol was used, demonstrating the easy use of PNA probes for in situ labeling assays.
Recently, Pellestor et al. (2003) experimented with PNA technology on human sperm. The adaptation of PNA technology to human spermatozoa constituted an interesting challenge because of the pecularities of sperm nuclei in terms of genomic compaction and accessibility of DNA sequences. To estimate and validate the efficiency of PNA labeling on human sperm, comparative estimates of disomies X, Y, and 1 were performed on sperm preparations from healthy subjects using multicolor FISH, PRINS, and PNA procedures in parallel. An equivalent quality of in situ nuclear labeling and similar disomy rates were obtained with the three methods. However, the hybridization timing of PNA probes (i.e., 45 min) was considerably shortened in comparison with FISH reaction, which requires an overnight hybridization to be efficiently completed on sperm preparations. The fast hybridization kinetics of PNAs on sperm were comparable to the kinetics of the PRINS reaction (20–30 min). This similarity might be due to the small size of both PNA probes and PRINS primers. These data pointed out the great potential of PNA probes for chromosomal screening on difficult biological material.
Finally, the PNA strategy has been used on isolated human oocytes, polar bodies, and blastomeres to assess the possibility of using PNA probes for preimplantation cytogenetic diagnosis (Paulasova et al. 2004). Using directly labeled satellite PNA probes for chromosomes 1, 4, 9, 16, 18, X, and Y, simple and sequential multicolor PNA labeling procedures were tested on 34 in vitro unfertilized oocytes and 23 blastomeres. The combined use of PNA and FISH was also investigated. Both rates and types of chromosomal abnormalities scored were in good agreement with results of previous FISH studies. This first use of PNA probes on isolated cells confirms the efficiency of PNA technology for in situ chromosomal analysis and demonstrates the feasibility of using PNAs on unique cells. This procedure could become an efficient complement to FISH for preimplantation genetic diagnosis because of its simplicity, its fast kinetics, and the high affinity of PNA probes.
Conclusion
PNAs have been implemented in research protocols and medical assays of increasing diagnostic value, adding both the sensitivity and the specificity of PNA probes to the standard procedures. Recent studies indicate that PNA probes have multiple advantages for the in situ analysis of nucleic acid sequences. Consequently, the PNA hybridization method may develop quickly within the field of in situ labeling methodology and become a powerful complement to FISH and PRINS for in situ chromosomal investigations.
New chemical modifications of the original PNA backbone may contribute to increasing the efficiency of PNA molecules and developing novel applications. Interesting new contributions of PNAs could come from the development of applications in the growing area of whole-genome analysis. The remarkable hybridization properties of PNA suggest that PNA oligomers may be efficiently incorporated into microarrays (Weiler et al. 1997). In association with different fluorochromes, short PNA sequences can constitute a new class of genomic biomarker for microarray platforms and contribute to the next challenge of extending microarray technology to the single-cell level and preimplantation diagnosis (Bermudez et al. 2004).
Another promising feature of PNAs might be linked to the development of in vivo fluorescence imaging. The capability of introducing fluorescent probes into living cells will allow deeper study of live gene expression and mRNA transfer (Tyagi and Kramer 1996). Because of their high in vivo stability and resistance to enzymes, PNA oligomers conjugated to cell-permeable peptides or liposomes have great potential for contributing to the future of noninvasive medical imaging.
Footnotes
Acknowledgements
This study was supported by European grant COPERNICUS 2 (Contract ICA-CT-2000–10012, proposal ICA2–1999–20007).