
Abstract
A surface-enhanced laser desorption/ionization time of flight (SELDI-TOF)-based ProteinChip System was used as a tool for rapid discovery and identification of protein patterns in serum that discriminate between trisomy 21 and unaffected pregnancies. We analyzed 24 serum samples from women carrying a trisomy 21 pregnancy and 32 with an unaffected pregnancy, ranging from 10.0 to 14.0 weeks of gestation. The resulting protein profiles were submitted to a clustering algorithm, a rule extraction, a rating, and a rule base construction step. For the generated combined rule base, the specificity and sensitivity for the prediction of a trisomy 21 pregnancy reach 97% and 91%, respectively.
Keywords
I
In the work presented here, we tried to find differentially expressed protein profiles by ProteinChip technology in maternal serum samples, which constitute an easy way to handle noninvasive first-trimester screening for trisomy 21 pregnancies.
Our study population consisted of 56 maternal serum samples, taken between weeks 10.0 and 14.0 of gestation. Twenty-four of these serum samples were from women carrying a fetus with trisomy 21 and 32 were from women carrying a fetus with a normal karyotype. The chromosomal status of the fetus was confirmed by amniocentesis or chorionic villous sampling. Untreated whole blood was collected and allowed to clot at room temperature. Serum was purified from blood by centrifugation at 1500 × g for 10 min, shock-frozen and stored at −80C. Serum samples were not subjected to more than two freeze-thaw cycles before the assay.
For analysis, the proteomic technique SELDI-MS (surface-enhanced laser desorption/ionization-mass spectrometry)-based ProteinChip technology was used (Melle et al. 2003). This technology makes use of affinity surfaces to retain proteins on the basis of their physicochemical characteristics, followed by direct analysis by time-of-flight mass spectrometry (TOF-MS). Proteins retained on chromatographic surfaces can be easily purified from contaminants such as buffer salts or detergents, thus eliminating the need for pre-separation techniques as are required for other MS techniques. For higher disintegration we used ProteinChip array types with two different surface chemistries: SAX2 as a strong anion exchanger and H50 as a reverse-phase or hydrophobic interaction array. For SAX2 arrays, the binding buffer consisted of 100 mM Tris (pH 8.5) containing 0.02% Triton-100. For H50 arrays, it was 30% acetonitrile and 0.1% triflouracetic acid in water. After equilibration, the buffer was removed and 3.5 μl of fresh buffer and 1.5 μl serum were added to each spot. The array was incubated in a humidity chamber for 90 min at 20C. Then each target was washed three times with 5 μl binding buffer, washed two times with 5 μl water to remove the buffer salts, and subsequently air-dried. Next, 2 × 0.5 μl saturated sinapinic acid dissolved in 50% acetonitrile containing 0.5% trifluoroacetic acid (all from Sigma; St Louis, MO) was applied to each spot. Mass analysis was performed in the ProteinChip PBSII instrument according to an automated data collection protocol. Spectra were analyzed using the ProteinChip software (version 3.1).
The resulting protein profiles were subjected to a cluster- and rule-based data mining algorithm. The data analysis process consists of a clustering step, a rule extraction and rating step, and a rule base construction step. All these steps, except for the clustering step, are supervised with respect to the given sample classification (e.g., trisomy 21 vs unaffected). The data were log 2-transformed before the normalization procedure and were then normalized to the median. The preprocessed protein expressions were then clustered into two clusters, “low expressed” and “high expressed,” for each peak using a modified fuzzy c-means algorithm (Bezdek 1981), in which samples with a protein expression below the lower cluster center were fully assigned to the cluster “low expressed” and samples with a protein expression above the higher cluster center were fully assigned to the cluster “high expressed.” The main objective of the next step, the rule extraction and rating step, is to find those features (peaks) that have the most similar membership distribution compared with the given sample class distribution. For this purpose, so-called rules are generated and rated by a statistically based rule rating measure introduced by Kiendl and Krabs (1989). Such rules can be written as follows: IF sample belongs to cluster “peak X high expressed,” THEN sample belongs to class “trisomy 21 pregnancy.” Rules with a rule rating measure greater than zero were then ranked in a rule list.
A small subset of rules out of the rule list can form a rule base that can be used for automatic classification of new patient samples. A rule base contains at least one rule for every possible classification outcome. To classify a new patient sample, the cluster memberships (condition part of the rules) of all rules out of the rule base that point to the same classification outcome (conclusion part of the rules) are added and the sample is assigned to the class with the highest sum. To construct a rule base that gives a good representation of the dataset investigated, all combinations of rules out of the rule lists were permuted and the rule base with the smallest classification error and the smallest number of rules was chosen. The rules contained in the chosen rule base can be considered to represent markers that can distinguish between the sample classes under investigation.
Analysis resulted in protein profiles for 56 patients for both ProteinChip Array surfaces. From these 56 profile sets, 24 were from women carrying a fetus with a trisomy 21 and 32 with a normal karyotype. Peaks were defined with a signal-to-noise ratio (S/N) greater than 5. Clusters were completed with estimated peaks added greater than 2 S/N, with a cluster mass window of 0.3% of mass. For the above-mentioned conditions in the range from 1.5 to 20 kD, 21 peaks for H50 arrays and 69 for SAX2 arrays could be found. In the range of 10 to 200 kD, 72 peaks for H50 arrays were found and 38 peaks for SAX2 arrays. The consideration of single peaks led to no sufficient discrimination between unaffected or trisomy 21 pregnancies. Moreover, we could not find a correlation of biomarkers within gestational age, although this is known for other serum markers (Wapner et al. 2003). The cluster- and rule-based data mining method described above revealed six peaks whose combination is relevant to distinguish between unaffected or trisomy 21 pregnancies (Table 1). These peaks are equally distributed between the two chip surfaces and between the criteria unaffected vs trisomy 21. They have a size ranging from 7.9 kD to 96.7 kD. The cluster borders are given in Table 2. One condition, for example, is if the expression at peak 34 is higher then 0.187 on H50 ProteinChip Array the sample belongs to a serum sample from a woman carrying a fetus with a normal karyotype. After combining all six generated rules (Table 1), it was possible to identify 24 samples of the trisomy 21 pregnancies, with a wrong prediction for two cases. Therefore, the calculated sensitivity is 91%. Of 32 unaffected pregnancies, one was predicted false-positive, resulting in a specificity of 97%. With this result, we reached an equal or higher sensitivity and specificity than ProteinChip System-based studies on cancer (Kozak et al. 2003; Won et al. 2003), also compared with other prenatal screening methods (Wapner et al. 2003) or to other testing based on fetal DNA in maternal serum (Lo 2000).
Combined rule base for H50 and SAX2 chip surfaces to distinguish between a trisomy 21 and an unaffected pregnancy a
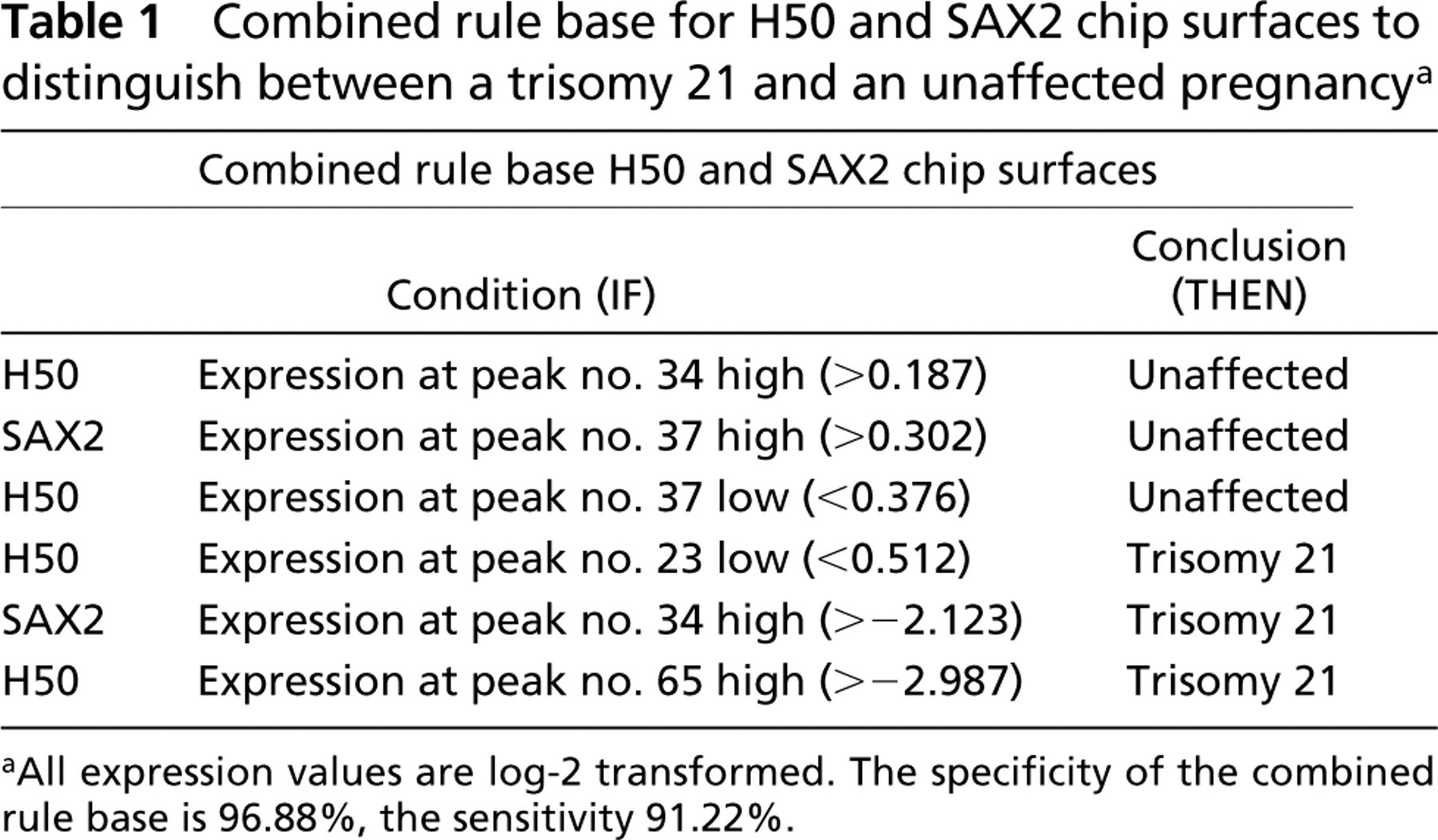
aAll expression values are log-2 transformed. The specificity of the combined rule base is 96.88%, the sensitivity 91.22%.
Cluster borders for rule base given in Table 1
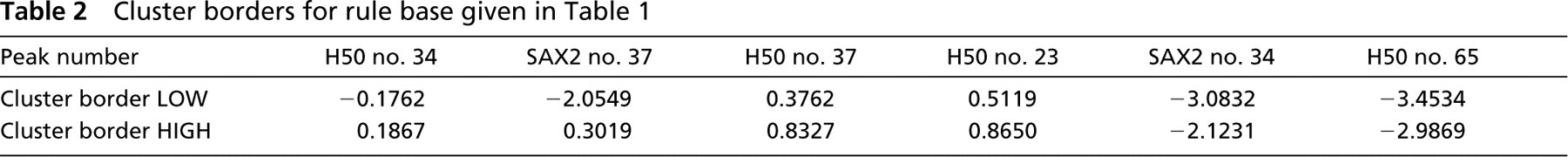
In conclusion, we have established a promising procedure combining ProteinChip technology and rule-based analysis that allows a first-trimester screening for trisomy 21 with a high sensitivity and specificity. The specific proteomic signatures described here, even without the knowledge of the respective proteins, can serve as an additional parameter for a more reliable detection of trisomy 21 pregnancies.
Footnotes
Acknowledgements
Supported by a grant from the German Federal Ministry of Education and Research (BMBF) and in part by the EU (ICA2-CT-2000–10012).
We thank Dr A. Kossakiewicz, Dr P. Kozlowski, Dr M. Pruggmayer, and Dr B. Eiben for their contributions of serum samples.