
Abstract
In the mid-1980s, two versions of Timm's original immersion sulfide silver method were published. The authors used immersion of tissue in a sulfide solution as opposed to Timm, who used immersion of tissue blocks in hydrogen sulfide-bubbled alcohol. The autometallography staining resulting from the “sulfide only immersion” was not particularly impressive, but the significance of this return to an old approach became obvious when Wenzel and co-workers presented their approach in connection with introduction by the Palmiter group of zinc transporter 3 (ZnT3). The Wenzel/Palmiter pictures are the first high-resolution, high-quality pictures taken from tissues in which free and loosely bound zinc ions have been captured in zinc-sulfur nanocrystals by immersion. The trick was to place formalin-fixed blocks of mouse brains in a solution containing 3% glutaraldehyde and 0.1% sodium sulfide, ingredients used for transcardial perfusion in the zinc-specific NeoTimm method. That the NeoTimm technique results in silver enhancement of zinc-sulfur nanocrystals has been proved by proton-induced X-ray multielement analyses (PIXE) and in vivo chelation with diethyldithiocarbamate (DEDTC). The aims of the present study were (a) to make the immersion-based capturing of zinc ions in zinc-sulfur nanocrystals work directly on sections and slices of fixed brain tissue, (b) to work out protocols that ensure zinc specificity and optimal quality of the staining, (c) to apply “immersion autometallography” (iZnSAMG) to other tissues that contain zinc-enriched (ZEN) cells, and (d) to make the immersion approach work on unfixed fresh tissue.
T
Physical development was used by photographers for artistic purposes to manipulate photos. The light-exposed photographic plate was depleted of its silver bromide crystals by thiosulfate, leaving only the tiny traces of metallic silver grains resulting from the light exposure. The invisible picture was then made visible by being developed in a “physical developer,” a misleading name based on the misconception that the silver amplification performed under these circumstances was different from the normal “chemical development” (Danscher 1996).
Since Timm's introduction of the concept that some metal sulfide accumulations could be silver-enhanced by physical development (Timm 1958,1962) many scientists have used his approach, have tried to improve the technique, and have added new metals to the family of nanocrystals that can be silver-enhanced (e.g., Haug 1967,1973,1974; Brunk and Brun 1972; Danscher 1981a, b; Sloviter 1990; Danscher and Møller-Madsen 1985; Zdolsek et al. 1993; Ross et al. 1996). In the early 1980s, Timm's original sulfide silver method was made zinc-specific (Danscher 1981c), and the selenium technique for in vivo creation of zinc-selenium nanocrystals in zinc-enriched (ZEN) neuronal terminals and for retrograde axonal tracing of ZEN neurons was introduced (Danscher 1982,1984a; Howell and Frederickson 1990). As a result of the growing understanding of the processes underlying silver enhancement, the term autometallography (AMG) was suggested to replace the term “physical development” when used in histochemistry (Danscher 1984b). This was particularly relevant because it was proved that metallic gold nanoparticles were the substrate for AMG silver enhancement in tissues from animals or humans that had been subjected to gold salts (Danscher 1981b). This finding led to the use of AMG for silver-enhancement of colloidal gold particles in immunohistochemistry (IHC) (Holgate et al. 1983) and for histochemical tracing of enzymes (Danscher and Nørgaard 1983).
Cassell and Brown (1984) and Tauck and Nadler (1985) described a version of Timm's sulfide silver method in which immersion in alcohol bubbled with hydrogen sulfide was substituted by the buffered sodium sulfide solution introduced by Haug for transcardial perfusion (for details see Haug 1973). This solution was also used in a 1% version for transcardial perfusion by Danscher and Zimmer (1978) and was finally corrected to 0.1% in the zinc ion-specific version of Timm's method, the so-called NeoTimm method (Danscher 1981c).
The 0.1% solution of buffered sodium sulfide was used to immerse human brain blocks by Franck et al. (1995) and Sutula et al. (1989). However, not until the Wenzel/Palmiter/Cole pictures from mouse brains, immersed in the combined glutaraldehyde-sodium sulfide solutions of the NeoTimm technique, did the benefit of this approach become appreciable (Wenzel et al. 1997; Cole et al. 1999; Franco-Pons et al. 2000).
The present study aims at establishing correctly performed immersion autometallography (iZnSAMG) as a highly specific tool for tracing zinc ions directly in tissue sections and to give easy-to-follow protocols of the technique and ways of controlling zinc specificity.
Materials and Methods
Thirty female mice of the BALB/Ca strain (Møllegaard Breeding Centre; Ejby, Denmark) and 30 rats of the Wistar strain were used. The animals were kept in plastic cages in a room with a 12-hr light/dark cycle at 21–22C and 50% humidity. They were fed Altromin No. 1324 (Spezialfutterwerke; Berlin, Germany) ad libitum and had free access to tap water. The study was undertaken in accordance with the Danish and University of Aarhus guidelines for animal welfare.
The animals were deeply anesthetized IP with Mebumal and sacrificed by decapitation or by transcardial perfusion with a fixative at a pressure of 120–130 mmHg. The perfusions were initiated by prewashing with an NaCl-heparin solution [9 ml 0.9% NaCl + 1 ml heparin (5000 IU/ml)] for 30 sec followed by transcardial perfusion with either (a) 250 ml 4% paraformaldehyde for 15 min, or (b) 250 ml fixative containing 1% paraformaldehyde and 1% glutaraldehyde for 15 min, or (c) 250 ml fixative containing 3% glutaraldehyde for 15 min, but without prewashing. All solutions were buffered with a 0.15 M Sørensen phosphate buffer.
Based on a multitude of experiments, the following procedures were found to be superior.
Immersion Procedure
One-, 2-, 4-, 12-, 16-, and 20-mm-thick tissue slices were cut by placing the brains in the “fast tissue slicer” (Histotech; Copenhagen, Denmark). The slices were immersed in a NeoTimm solution (NTS), i.e., 0.1% sodium sulfide and 3% glutaraldehyde in a 0.1 M Sørensen phosphate buffer, pH 7.4. The immersion jars were placed on a shaker and kept at 4C. Three days later the slices were carefully rinsed twice in 0.1 M phosphate buffer for 10 min. The fixed slices were further processed with or without AMG development.
Non-developed NTS Slices
Cryostat Sections. The slices were placed in a 30% solution of sucrose until they sank to the bottom of the glass. They were shortly thereafter frozen with CO2 on a cryostat stage, placed in a cryostat, and allowed to increase in temperature to −17C. Thirty-μm-thick sections were cut, placed on Farmer cleaned glass slides and AMG developed. After development the sections were counter-stained with a 0.1% aqueous toluidine blue solution (pH 4.0), dehydrated in alcohol to xylene, and ultimately embedded in DEPEX and covered with a coverglass.
Paraffin Sections. The slices were embedded in paraffin while checking that the temperature during the embedding did not exceed 45C, cut into 5-μm-thick sections, and placed on glass slides. These were AMG developed, counterstained with TB, and mounted in DEPEX.
Epon Sections. The small blocks cut out from the slices were embedded in Epon while keeping the polymerization temperature below 45C. From the Epon blocks, 3-μm sections were cut, placed on glass slides, and AMG developed. One of the three sections on each glass slide was counterstained with toluidine blue. After LM analysis, sections to be analyzed in the electron microscope were reembedded on top of an Epon block and from these preparations ultrathin sections were cut, placed on a grid, and counterstained with uranyl citrate and lead acetate.
AMG-developed NTS Slices
Paraffin Sections. The slices were embedded in paraffin without temperature control and 5-μm-thick sections were cut, placed on glass slides, counterstained with toluidine blue, and mounted in DEPEX.
Epon Sections. The areas to be analyzed in the EM were dissected from the AMG-developed slices and these small blocks of tissue were placed in a vial in which they were fixed in 0.1% osmium tetroxide for 30 min. After proper rinsing in distilled water, the sections were embedded in Epon without temperature control. From these Epon blocks, 3-μm-thick sections were cut and analyzed in the LM. The sections to be analyzed were then reembedded on top of a blank Epon block, and after proper trimming ultrathin sections were cut and stained
The AMG Silver Lactate Enhancement Developer (pH 3.8)
The AMG developer consists of a 60-ml gum arabic solution and 10 ml sodium citrate buffer (25.5 g of citric acid 1 H2O + 23.5 g sodium citrate 2 H2O to 100 ml distilled water), 15 ml reductor (0.85 g of hydroquinone dissolved in 15 ml distilled water at 40C), and a 15-ml solution containing silver ions (0.12 g silver lactate in 15 ml distilled water at 40C) added immediately before use while thoroughly stirring the AMG solution (Danscher 1981a; Stoltenberg and Danscher 2000).
AMG Development of Tissue Sections Placed on Glass Slides
The glass slides were placed in Farmer-cleaned jars, poured with the AMG developer and placed in a water bath at 26C on an electric device that shook the jars gently. The entire set-up was prepared in plain daylight on the lab bench but was covered by a dark hood throughout the actual development. After 60 min the AMG development was stopped by replacing the developer with a 5% sodium thiosulfate solution for 10 min (AMG stop bath). The jars were then placed under gently running ion-exchange water for 5 min.
If the tissue sections reveal a high background staining, two approaches have been worked out: (a) The glass slides are dipped in a 0.5% solution of gelatin and allowed to dry before development. After AMG the jars are placed under 45C warm tapwater for 15 min before they are rinsed in distilled water, counterstained, and coverslipped. (b) The glass slides are dipped in Farmer solution for 10–30 sec, rinsed in distilled water, counterstained, and coverslipped (for details see Danscher 1996).
AMG Development of NTS Slices
The slices were placed in Farmer-cleaned jars and poured with the AMG developer. The jars were placed in a water bath at 26C and covered by a light-tight hood. After 60 min the development was stopped by replacing the developer with 5% sodium thiosulfate. After 10 min the slices were rinsed carefully for 5 min in several washes of distilled water.
Human Tissue
Human tissues obtained from autopsies or biopsies were fixed in 4% buffered formalin for 2 hr before being immersed in the NTS for 3 days. Sections for LM and EM analysis were performed as described above. If the tissue blocks are less than 2 mm in diameter, they can be placed in the NTS without previous cutting.
Alternative Ways of Performing iZnSAMG
It is feasible to freeze and cut whole blocks of fresh or perfusion-fixed tissue, as illustrated with black arrows in Figure 3. Following this methodological pathway, glass-mounted sections subjected to the NTS leave the option of applying other histochemical techniques on NTS-free alternating glass slides of parallel series of sections from the same tissue block, e.g., iZnSAMG and IHC detection of zinc transporters: (a) fixed tissue blocks were removed, transferred to sucrose, frozen and cut as described above; (b) in Figure 3B, unfixed tissue blocks are removed, frozen with CO2 immediately after removal, and cut on a cryostat. The sections are placed on glass slides and immersed in 3% buffered glutaraldehyde or 4% buffered paraformaldehyde for 2 hr before immersion in NTS.
It is possible to thaw blocks of tissue and to use 1-2-mm-thick slices for the immersion AMG.
Additional Comments and Observations
Immersion in NTS in which sodium sulfide is replaced with sodium selenite was not successful.
Results
Slice Thickness
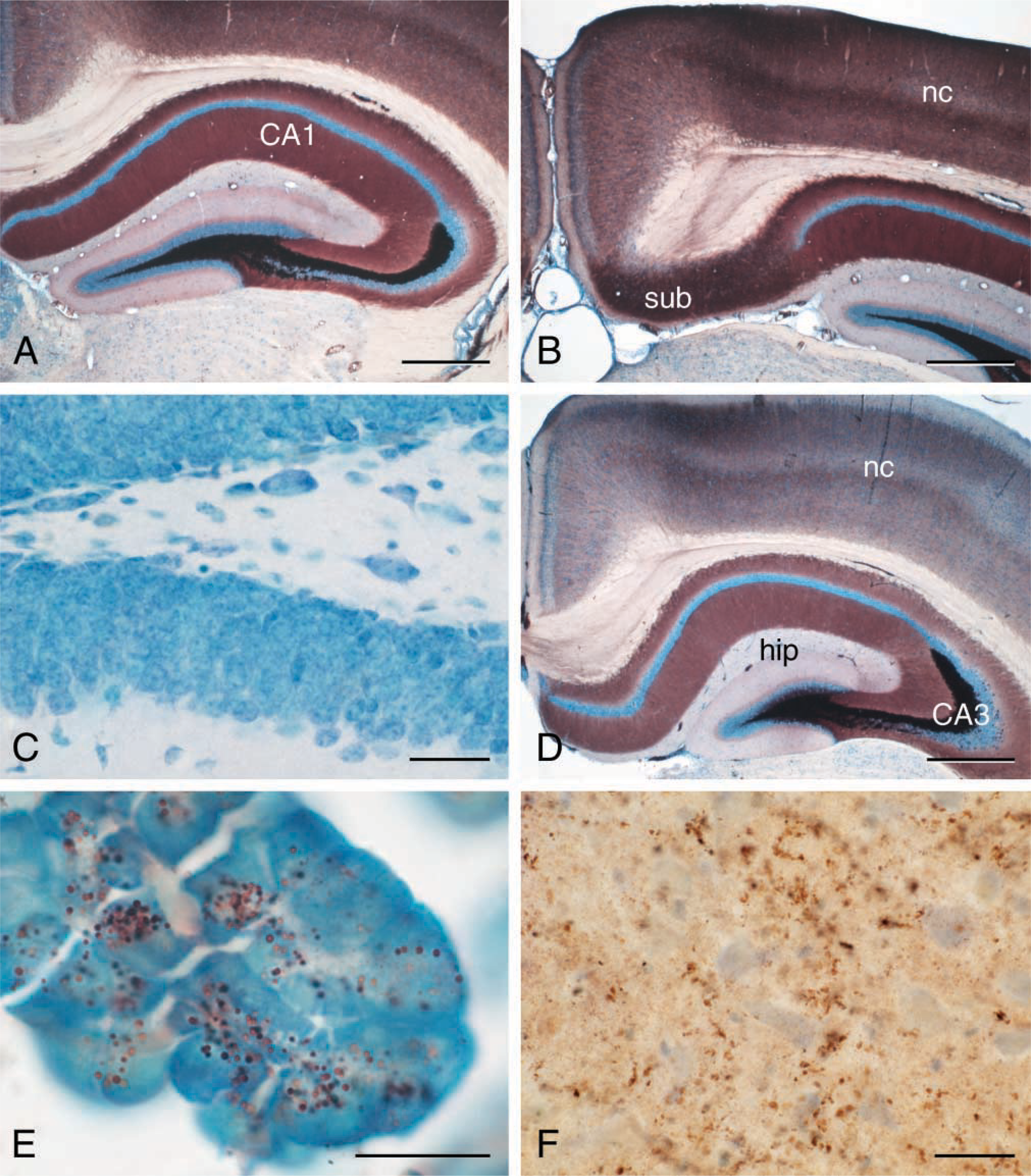
We have found that tissue slices up to 2 mm thick are superior for the iZnSAMG technique. It is of no or little significance whether the tissue has been fixed or not before being cut or vibratomized. However, paraform-aldehyde-prefixed tissue might result in slightly more intense staining of regions with few or small ZEN terminals (Figures 1A, 1B, and 1D). Unspecific staining, i.e., non-zinc-sulfur nanocrystal-based staining, might show up in vessels and myelin of the paraformaldehyde-fixed sections. If for one reason or another it is necessary to expose tissue blocks that are thicker than 2 mm to NTS, sections from such blocks must be AMG-developed for more than 60 min, and the quality of the staining is unpredictable and never as good as sections from blocks/slices ×2 mm thick.
The Optimal NTS Immersion
We have found that NTS immersion for 72 hr of tissue blocks with the ideal dimensions, i.e., ×2 mm thick, gives rise to the best quality of both iZnSAMG staining and tissue sections. It was equally obvious from our experiments that this period of immersion is to be chosen also for blocks thicker than 2 mm. Periods of immersion longer than 3 days will cause false AMG staining in the outer part of the block, and shorter periods will result in incomplete capturing of all zinc ions in nanocrystals in the deeper part of the block.
Electron Microscopy
The excellent quality of the technique at the ultra-structural level is demonstrated in Figure 2.
(
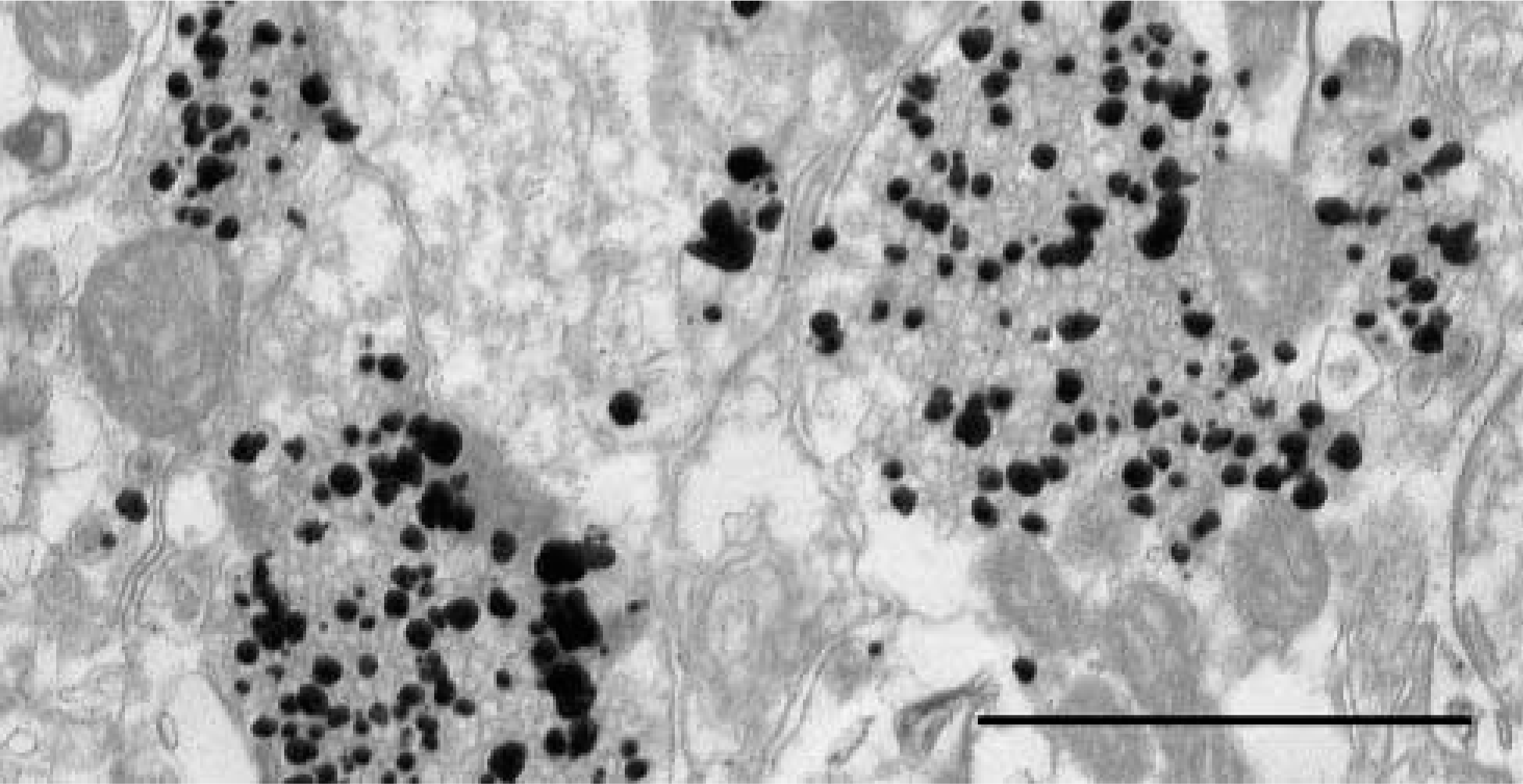
Electron micrograph of ZEN terminals in mouse neocortex. Note the many non-ZEN synaptic vesicles. Bar = 1 μm.
NTS-based iZnSAMG Specificity for Zinc
The above optimal condition for iZnSAMG has been tested for zinc specificity in three different ways: (a) animals have been treated with 1000 mg of the low-toxic zinc chelator diethyldithiocarbamate (DEDTC) 1 hr before sacrifice, and the brains and other zinc ion- containing tissues have been treated in the above optimal standard way—in sections from these sources we found no staining at all (Figure 1c); (b) 2-mm slices placed in DEDTC 1 hr before transferring them to the NTS solution likewise resulted in a total lack of iZnSAMG staining; (c) sections from tissue slices not exposed to NTS immersion but AMG-developed were devoid of staining.
Human Brain
Figure 1F demonstrates ZEN terminals in neocortex from a brain autopsy. The procedures are summarized in Figure 3.
iZnSAMG Staining of Zinc Ions in Zinc-enriched Cells
The iZnSAMG technique works well on other types of ZEN cells, including pancreas (Figure 1E), gut, prostate, liver, and kidney.
Discussion
The possibility of tracing loosely bound or free zinc ions in tissue sections by iZnSAMG is good news. Until recently this could be done only by using chelating fluorescent agents such as TSQ (Frederickson et al. 1987) and zinquin (Zalewski et al. 1993) or by exposing the sections to H2S gas, followed by autometallographic silver enhancement of the created zinc-sulfur nanocrystals (Jaarsma and Korf 1990; Danscher et al. 1997). However, the results obtained by immersing tissue blocks of brain tissue from mice and humans in the NTS (Wenzel et al. 1997; Franco-Pons et al. 2000) changed this situation completely. The procedure of creating zinc-sulfur nanocrystals in cellular compartments that contain loosely bound or free zinc ions can be performed on most tissue sections, i.e., tissue sections on glass slides, cell cultures, or samples of cells, whether fixed or unfixed, the only limitation being that the zinc ions are not removed by prolonged fixation, chelation, or in any other way expelled from the cells. When the cell cultures are not grown on small glass slides or when the cell samples cannot be placed on glass slides as a smear, the cells are poured with NTS and allowed to stay for a couple of hours. The cells are then centrifuged to the bottom of the vial and washed a couple of times with a phosphate buffer before being centrifuged again and AMG developer added. After development and careful rinsing in distilled water, the pellet can either be embedded in Epon or smeared on a glass slide.
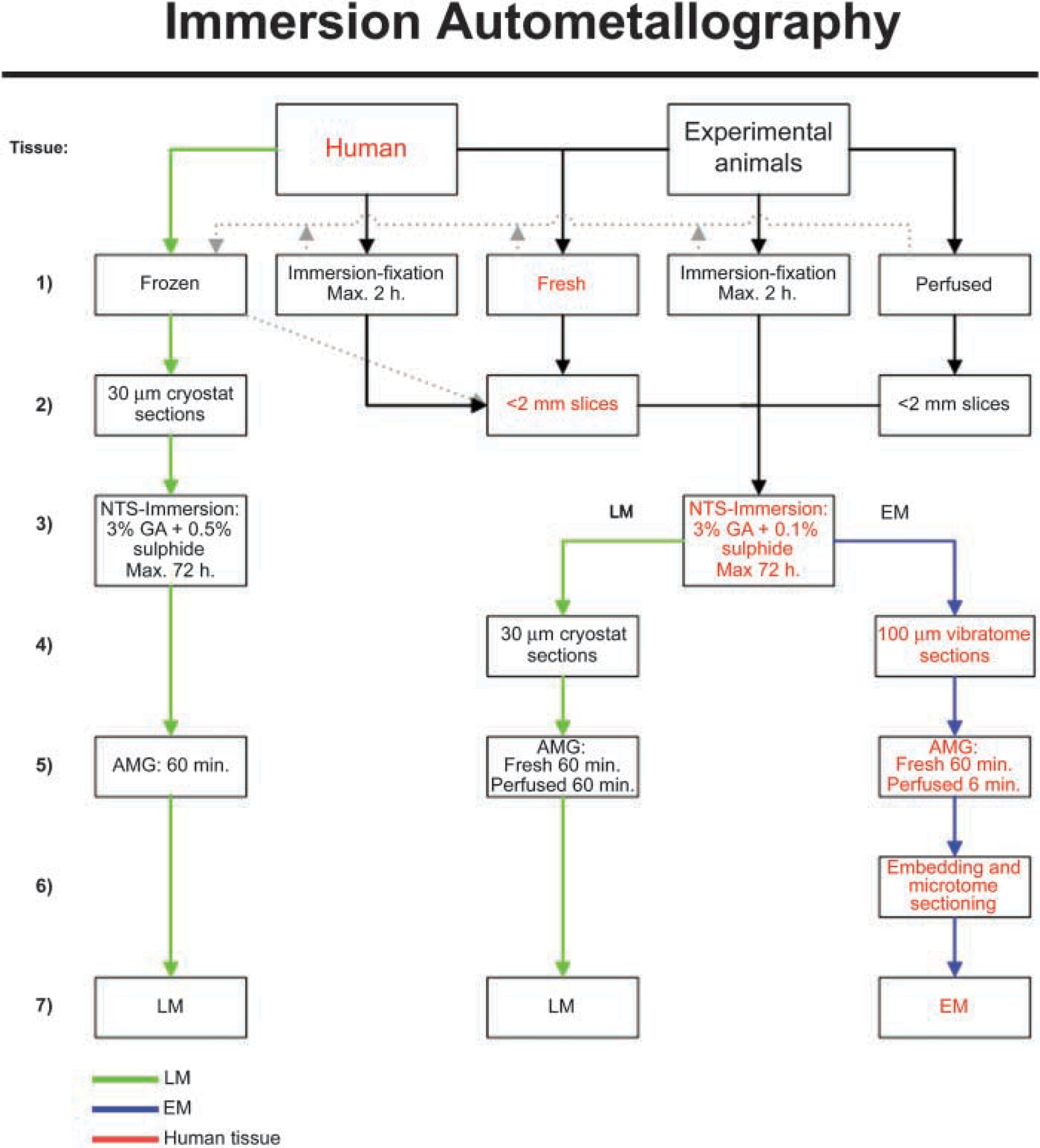
Schematic illustration the different ways of performing immersion autometallography.
The approach is easy to control for specificity by chelation, e.g., by treating live animals or the cultures/ cell samples with diethyldithiocarbamate (DEDTC), whereby zinc ions are bound in vivo as zinc DEDTC, or by placing the tissue slices directly into solutions of DEDTC or TSQ before the NTS immersion.
The possibility of creating the more stable zincselenium nanocrystals by immersion in a solution of sodium selenide and glutaraldehyde by using the more stable selenide ions instead of sulfide ions, i.e., creating zinc-selenium (ZnSe) nanocrystals, has been tested and found not to be an option. We do not know why sodium selenide creates zinc-selenium nanocrystals when applied in vivo or by transcardial perfusion (Danscher 1982,1984b,c,d; Slomianka et al. 1990), but is of no value for immersion capturing of zinc ions.
We recommend the following AMG approaches for specific histochemical tracing of zinc ions in neuronal terminals: (a) transcardial perfusion with 0.1% sodium sulfide and phosphate-buffered 3% glutaraldehyde (the NeoTimm method); (b) immersion of up to 2-mm slices of tissue in the NeoTimm solution (immersion autometallography); (c) in vivo treatment with sodium selenite systemically, i.e., by IV or IP application, or locally with sodium selenide, e.g., by intracerebral injection, and survival times from 1 to 2 hr for tracing ZEN terminals in CNS and PNS; (d) systemic or local injection of, respectively, sodium selenite and selenide and survival times from 18 to 36 hr for tracing ZEN fiber tracts by retrograde axonal loading of ZEN somata.
Footnotes
Acknowledgements
Supported by The Danish Medical Research Council, Aase & Ejnar Danielsens Fond, Aarhus University Research Foundation, and the Lundbeck, Leo, Beckett, Gangsted, and Novo Nordic Foundations.
We gratefully acknowledge the skillful technical assistance of Ms Helene Andersen, Mr A. Meier, Ms H. Mikkelsen, Ms Lene Munkøe, Mr T.A. Nielsen, Ms M. Sand, and Ms K. Wiedemann.