
Abstract
We present a new technique that allows zinc ions in synaptic and secretory vesicles of biopsy and early autopsy material (>2 hr post mortem) to be transformed to nanometer-sized zinc sulfide crystal lattices for subsequent autometallographic (AMG) development. Human brain biopsies, or other tissue samples containing zinc-enriched (ZEN) cells, are frozen in liquid nitrogen or by CO2 gas immediately after removal. The tissue blocks are cut in a cryostat and the sections placed on glass slides. The slides are transferred to an H2S exposure chamber placed in a −15C freezer. After 1–24 hr of gas exposure the sections are removed from the chamber, fixed while thawing, and dehydrated. The sections are then exposed to an AMG developer. AMG causes silver enhancement of zinc sulfide crystal lattices created in the tissues through the H2S exposure, making them visible. It is imperative that the tissues are frozen instantaneously after removal, because loosely bound or free zinc ions start leaving their vesicular compartment soon after death. The AMG technique can, despite inadequate fixation and damage to the tissue caused by freezing, also be used to trace zinc ions at ultrastructural levels, and it is demonstrated that zinc ions in the human neocortex are located in synaptic vesicles. In the few human biopsies analyzed thus far, the light microscopic pattern created by the silver-enhanced ZEN terminals resembles that seen in the neocortex of rat brain. The technique has been applied to cryostat sections from neocortex biopsies of five individuals undergoing brain surgery. Biopsies from three patients resulted in satisfactory AMG-stained sections. Rat brains removed and frozen immediately after decapitation constituted the material on which the present technique was developed. Such material results in an almost uniform high quality of staining, and we found that unexposed sections can be stored for at least 5 months at −80C without ensuing significant loss of AMG staining intensity.
Keywords
T
Since Timm's original “sulphide silver staining” for visualization of the “Maske zinc pool” was introduced in 1958, it has been a goal for many researchers to develop a Timm technique that could be performed on tissue sections, i.e., where the chemical binding of zinc ions to sulfide could take place on tissue sections placed on glass slides.
The original Timm method used fixation of the tissue block with alcohol bubbled with H2S gas. This technique was, however, not specific for zinc, most probably because the alcoholic denaturation of the tissue proteins caused release of copper and possibly other metals. A breakthrough in the literature was Haug's introduction of transcardial perfusion with a 1% sodium sulfide solution. By using cryostat sections of brains from the perfused rats, Haug was able to obtain a satisfactory staining (for details see Haug 1973). However, because of the excessively high levels of sulfide ions he introduced a factor of uncertainty. A surplus of sulfide ions causes adherence of some of them to molecules in the tissue. When such sulfide-loaded sections are exposed to the autometallographic (AMG) developer, the extra pool of sulfide ions will bind silver ions from the AMG developer, creating small silver sulfide crystal lattices that additionally will be silver-amplified by the developer. Because only certain molecules (possibly –SH-containing proteins) can bind sulfide ions in this way, the non-Zn AMG staining reveals an extremely orderly distribution and can easily be misinterpreted as being a genuine pattern of ZEN terminals (see Danscher 1993). This problem was solved in the NeoTimm method (Danscher 1981) which also included a newly composed AMG developer together with other technical improvements aimed to reduce the unspecific background staining to a minimum. The NeoTimm method made Timm's sulfide silver method a tool for specific demonstration of ZEN vesicles.
Several attempts have been made to apply Timm's method to tissue sections using either H2S gas exposure or exposure to sulfide ions from a solution of sodium sulfide (Brunk et al. 1968; Chafetz 1986; Jaarsma and Korff 1990). Jaarsma and Korff introduced a technique in which thawed cryostat sections from rat brains and human autopsy material were exposed to H2S gas for several hours. When applied to cryostat sections from human autopsy material, the Jaarsma–Korff method proved to be unsuccessful in our hands, but with rat brains we found the method successful in one out of 10 trials. When successful, the staining pattern appears to be of good quality at low magnification but blurred at higher magnification. The hazy stain suggests that zinc ions have drifted away from their original position in the synaptic vesicles. Such a drift of zinc had been observed in fixed material by Kemp and Danscher in 1979, and we therefore decided to keep the rat brain sections frozen until the moment at which the vesicular zinc ions were converted into zinc sulfide crystal lattices in situ. In this way, leakage of zinc ions from the synaptic vesicles caused by thawing was circumvented. The approach was found to be valuable and was applied to biopsy material frozen immediately after removal from humans undergoing surgical treatment.
Materials and Methods
Cryostat sections of biopsies from the neocortex of five individuals undergoing brain surgery and brains from 21 rats removed and frozen immediately after decapitation were used to work out the present technique.
Construction of the “Sulfidation Device”
Hydrogen sulfide is highly toxic and very reactive, and the gas must therefore be handled with a totally closed system made of inert materials. When expelled from the exposure system, e.g., by nitrogen gas, the H2S gas must bubble through a hypochlorite solution where it will be chemically destroyed (sulfide is oxidized to free sulfur).
The H2S exposure system was constructed de novo using three stainless steel (quality type 316) cylinders (100 mm in diameter and 500 mm in length) closed at both ends with lids of aluminium placed on o-rings of butyl rubber greased with silicon vacuum grease and kept in place by wing screws. The cylinders are bound together and placed in a standard freezing cabinet (Gram FS type FS100) with a thermostat ranging from −10 to −20C.
Outside the freezing cabinet is placed a low-pressure gas cylinder containing hydrogen sulfide (AGA, purity 2.8) and a high-pressure cylinder containing nitrogen (AGA, “Plus” quality). The regulator on the hydrogen sulfide gas cylinder is made of stainless steel with movable parts of teflon (AGA type TDR715S).
The gas cylinders are connected with the three gas chambers inside the freezing cabinet through a system of 1/8″ stainless steel (316) high-pressure tubes and fittings known from high-pressure chromatography. This system is made with gas valves to keep the hydrogen sulfide and nitrogen gas cylinders isolated from each other. Otherwise the high-pressure nitrogen may flow into the low-pressure hydrogen sulfide gas cylinder by accident. In the freezing cabinet, 3 m of the tube is wound up as a spiral and placed in a plastic container filled with water ice. This part of the arrangement ensures that the gas temperature is reduced to a temperature close to the freezing cabinet.
The outlet from the gas chambers is likewise made with 1/8″ tubes connected to a common output into an empty washing bottle, followed by two washing bottles each containing 200 ml of a 25% sodium hypochlorite solution. The bottles are closed tightly with silicon vacuum grease and plastic rings and placed in a fume cupboard. When the nitrogen-expelled H2S gas passes through the hypochlorite solution, the sulfide is reduced to free sulfur.
Handling of Tissue Sections
Biopsies from human patients were frozen in liquid nitrogen immediately after removal in connection with surgical intervention. Likewise, brain and other organs removed from deeply anesthetized or decapitated Wistar rats were frozen on removal. These biopsies and tissue blocks were kept at −80C.
The frozen tissue blocks were glued to a cryostat stage by Tissue-Tek OCT 4583 Compound (Miles; Elkhart, IN), placed in a cryostat, and allowed to increase in temperature to −17C. Then 30-μm-thick sections were cut and mounted on Farmer cleaned (see below for explanation) glass slides. The sections were thawed for a few seconds to adhere to the slide and then stored at −80C until moved to the sulfidation chamber. The sections are placed in the chamber for 30 min before being flushed with H2S gas for 2 min, followed by 5 min of intermission and another 2 min of H2S flushing. Gas flow is adjusted to a level at which vivid bubbling is seen in the washing bottles.
The exposure time in the now H2S-filled chamber varied from 1 to 24 hr. In most cases an exposure period of 6–8 hr was found to be optimal, but in order not to miss any traces of zinc ions in the sections it is advisable to expose initially for 24 hr.
After H2S exposure, the chamber was flushed with nitrogen gas for 10 min. Then the chamber was opened and the cradles removed and placed in 70% alcohol for 30 min. Later, the sections were rehydrated and finally dipped in a 0.5% gelatin solution.
Because the AMG process is very sensitive, just 1 gold atom, 3–6 silver atoms or zinc sulfide molecules can catalyze silver precipitation on their surfaces (James 1977; Danscher 1981,1996), all traces of AMG catalysts must be removed from glassware and other utensils coming in contact with the AMG developer. This is done by rinsing with Farmer's solution made of one part 10% potassium ferricyanide (oxidizing agent) and nine parts 10% sodium thiosulfate (removes silver by complex binding) (Farmer 1884), followed by careful rinsing in distilled water.
Preparation of the AMG Solution
1. Protecting colloid. One kilogram of natural gum arabic resin drops from African acacia trees (Bidinger; Aarhus, Denmark) is dissolved in 2 liters of de-ionized water by intermittent stirring over 5 days. The solution is then filtered through layers of gauze to remove various impurities. The resulting colloid is then placed in plastic flasks of suitable sizes and placed at −25C. Made in this way the gum arabic solution can be stored for more than a year.
2. Citrate buffer. 25.5 g citric acid, 1 H2O, 23.5 g sodium citrate, 2 H2O, distilled water 100 ml.
3. Reducing agent, 0.85 g hydroquinone in 15 ml heated distilled water.
4. Silver ion supply. 0.11 g silver lactate in 15 ml 40C distilled water. The vial should be wrapped in a light-proof foil.
5. Mixing of the AMG ingredients. 60 ml protecting colloid (1), 10 ml citrate buffer (2), and 15 ml reducing agent (3) are carefully mixed, and finally 15 ml of the silver lactate solution (4) is added just before development.
AMG Development for Light Microscopic Analysis
In daylight the AMG developer was poured into Farmer-rinsed jars and placed at 26C in a waterbath equipped with a shaking device and placed in a light-tight box. Fifteen minutes later the slides were placed in the jars and the lid was replaced (Danscher 1996).
After 60 min of development the AMG process was stopped by replacing the developer in the jars with a 5% thiosulfate solution. After 10 min the jars were placed under running 40C tapwater for 20 min to remove the gelatin coat and then dipped twice in distilled water. Every second slide was immersed in a 2% Farmer solution for 30 sec and carefully rinsed in distilled water. All sections were finally counterstained with toluidine blue. After rinsing and dehydration the sections were mounted in DePex mounting medium (BDH Laboratory Supplies; Poole, UK).
A test slide with cryostat sections from a rat brain taken from an animal transcardially perfused with 0.1% sodium sulfide for 7 min (the NeoTimm method; Danscher 1981) was included in each jar as a control of successful AMG development.
AMG Development for Ultrastructural Analysis
Cryostat sections to be analyzed in the electron microscope were cut 50–70 μm thick and placed in a small (volume 8 ml) teflon bowl kept at −17C. The bowl was transferred to the gas chamber. After H2S exposure it was filled with cold 3% glutaraldehyde in a 0.1 M phosphate solution. The sections were allowed to thaw for 30 min in this solution at room temperature, then rinsed in distilled water three times, submerged in AMG, and developed at 26C for 60 min in the AMG device described above. After 10 min in the stopbath the sections were analyzed in a stereo microscope, and selected parts of the sections to be analyzed in the EM were cut out, fixed in a 1% osmium tetroxide solution in 0.1 M phosphate buffer (pH 7.4), and embedded in Epon. Two-μm-thick semithin sections were cut and, after light microscopic evaluation, re-embedded on top of a blank Epon block. From this preparation ultrathin sections were cut and stained with uranyl acetate and lead citrate (Danscher 1996).
Effect of Long-term Low-temperature Storage of Tissue Sections
Six rats were sacrificed by decapitation, the brains removed quickly, frozen in CO2, and stored at − 17C. The next day one of the hemispheres of each brain was placed in a cryostat and 30-μm sections were cut as described previously. The slides and remaining hemisphere from each animal were wrapped with plastic freezer film and stored at −80C.
After 1, 2, 3, 4, 5, and 6 months, sections were cut from a hemisphere and together with the previously prepared sections were exposed to hydrogen sulfide in the gas chamber and AMG developed as described above.
Results and Comments
The period of time necessary to convert all the zinc ions in a certain batch of sections to zinc sulfide crystal lattices in the H2S chamber was found to depend solely on the temperature in the chamber. The 30-μm-thick cryostat sections of rat brain used as a test tissue in the present study showed creation of zinc sulfide crystal lattices after 10 min at −15C, and 24-hr exposure resulted in an AMG staining that technically was of almost equally high quality to that of transcardially perfused rat brains (Figures 1a and 1b). The temperature of the chamber should always be below −5C because the H2S gas exposure at higher temperatures was found to be harmful to tissue morphology and to cause creation of nonspecific AMG grains. The reason for these problems might be a high pH in the tissue created by the H2S gas.
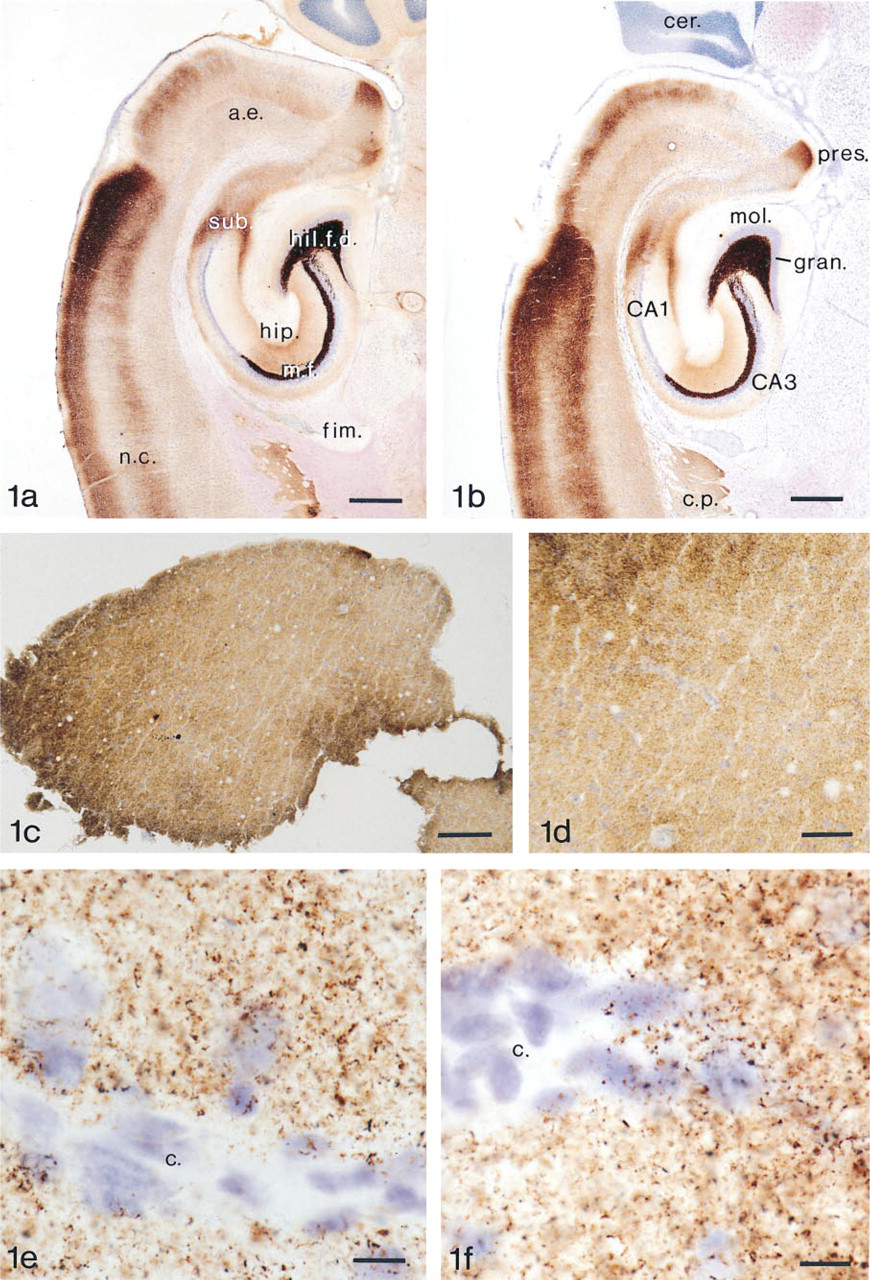
AMGZnS-H2S. Thirty-μm-thick section of rat hippocampus. The animal was sacrificed by decapitation. After removal the brain was frozen in liquid nitrogen. Two days later the brain was placed in a cryostat (Dittes Duspiva) at −17C and glued to the tissue holder. Thirty-μm-thick sections were cut and placed on glass slides. These were placed in the H2S gas chamber for 24 hr. The sections were AMG-developed and counterstained with toluidine blue. Bar = 625 μm. (
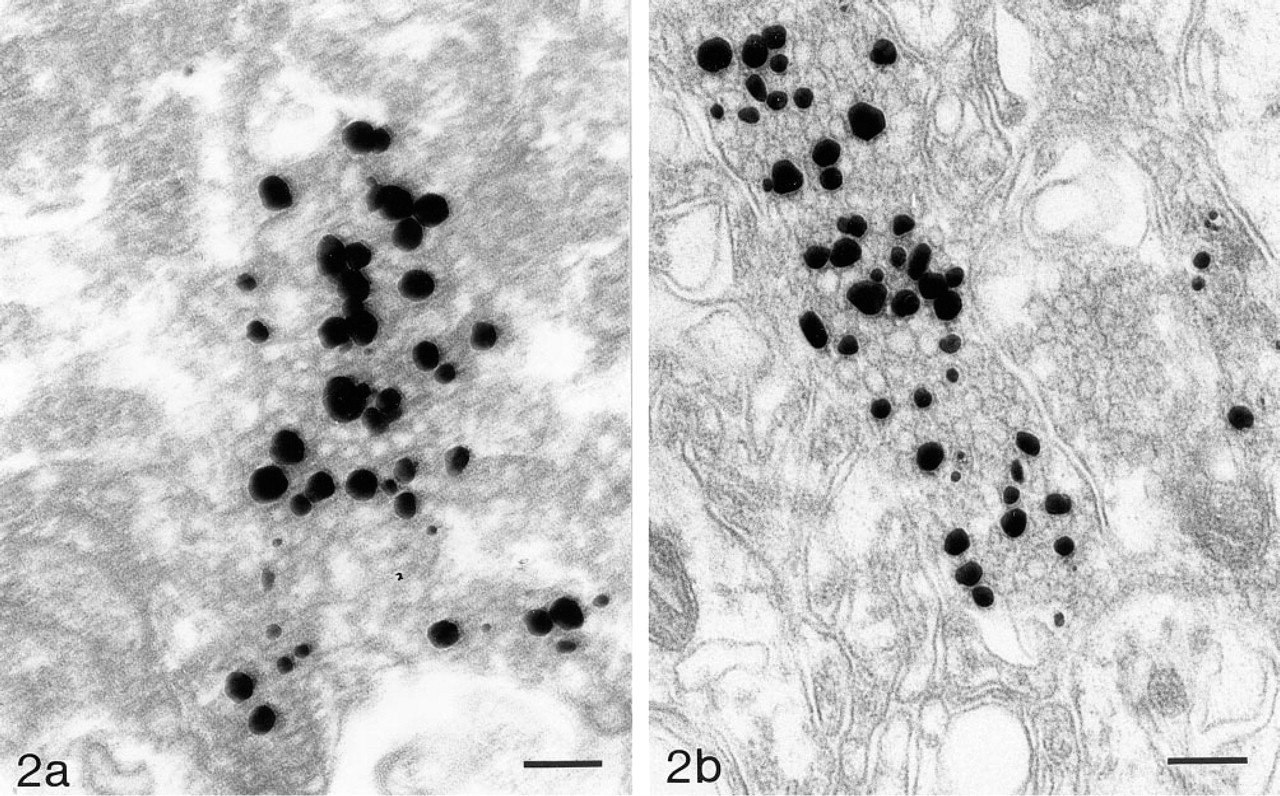
Electron micrograph of ZEN terminal in human neocortex. The specimen was frozen in liquid nitrogen immediately after removal, cut in a cryostat, exposed to H2S gas at −25C, fixed in glutaraldehyde, AMG-developed and embedded in epon. Bar = 156 nm. (
The time scale for leakage of zinc ions from their vesicular environment was analyzed by sacrificing rats and keeping them at room temperature for 2, 4, or 6 hr before the brains were removed and frozen. A substantial decline in staining intensity was found after 2 hr, and after 6 hr the diffusion leakage and chemical binding of zinc ions to molecules in the cytosol reduced the intensity of the AMG staining, which also became blurred and was of low diagnostic value. Only in the hippocampal mossy fiber system and the amygdaloid complex could the AMG staining still be recognized as bouton related, possibly because of the “autoTimm” effect (Danscher and Zimmer 1978) or because of the high density of ZEN vesicles in these areas (Pérez-Clausell and Danscher 1986).
The human biopsy material used in this study was frozen in liquid nitrogen immediately after removal (Figures 1c–1c). The quality was satisfactory in three of the five cases, and the staining pattern created by the ZEN terminals was almost identical to that found in rat neocortex (Figure 1e, Homo; Figure 1f, rat). Ultrastructural analysis of ultrathin sections from the 50-μm-thick cryostat sections showed that the zinc ions are located in the synaptic vesicles of ZEN terminals in the human neocortex (Figure 2a). Because of freezing of the tissue the quality of the electron microscopic sections is far from satisfactory, and we are presently trying to improve the technique to obtain tissue preservation closer to that seen in perfused experimental animals (Figure 2b).
Some of the human brain biopsies did not stain at all. This is most probably because they have been deprived of oxygen, e.g., by arteries being clamped or heated (burned) during surgery.
To ensure that the zinc pattern demonstrated by the H2S gas technique was caused by zinc ions, deeply anesthetized rats were treated
From previous work it is known that chelation with dithizone results in the same lack of staining and that proton activation (PIXE) multielement analysis (Kemp and Danscher 1979) of dithizonate extracted from such brains by carbon tetrachloride revealed zinc (Danscher et al. 1985).
The long-term storage of tissue blocks and sections at −80C did not influence the staining pattern, although a minor reduction in intensity was seen after 6 months. A difference between storage as tissue blocks or sections was not noticeable, but the risk for freeze-drying effects is higher for the sections.
Discussion
The pool of zinc ions that can be silver-amplified by AMG is localized in a subclass of synaptic vesicles (Danscher 1984; Friedman and Price 1984; Pérez-Clausell and Danscher 1985), and is released in a calcium-dependent way into the synaptic cleft (Assaf and Chung 1984; Howell et al. 1984; Pérez-Clausell and Danscher 1986). Moreover, it has been shown in vitro that zinc ions influence postsynaptic receptor material such as NMDA and GABA (Rassendrew et al. 1990; Xie and Smart 1993). Because zinc is present in cells from all tissues and in many different compartments of the cells (Frederickson 1989; Frederickson and Danscher 1990), the terms “zinc-containing” or even “vesicular zinc” are not adequate expressions for this particular type of neuron. The term “zinc-enriched,” abbreviated ZEN, has been suggested for this particular kind of neuron (Danscher 1994) to indicate that they are enriched with an additional zinc pool of loosely bound or free zinc ions. Most if not all ZEN neurons in the brain are excitatory and most likely glutaminergic (Frederickson et al. 1990).
The pool of zinc ions contained in synaptic vesicles of ZEN neurons is quite stable after transformation to zinc sulfide as long as the sections are not heated above 45C and the pH is kept neutral to weakly basic (Danscher 1981). However, if the zinc ions are not bound chemically, by either sulfide ions or selenide ions (Danscher 1982), they will diffuse quite rapidly from the synaptic vesicle compartment post mortem. Most likely the freed zinc ions bind to molecules in the cytoplasm, in the membrane, or outside the neurons, irrespective of whether the tissue is fixed or is left untreated. The only way to keep the ions in situ and available for crystal formation with sulfide or selenide ions is therefore to expose the tissue in vivo or by perfusion to S-2 or Se-2 ions, or to freeze the tissue so that the zinc ions remain in situ and chemically unchanged until they can be transferred to zinc sulfide crystals by being exposed to H2S while the tissue is still frozen.
Timm introduced the “sulphide silver method” in 1958, and until now a successful technique for Timm staining of tissue sections has not been published. As mentioned earlier, the use of H2S gas has been suggested by several authors (Brunk et al. 1968; Chafetz 1986; Jaarsma and Korff 1990), but the resulting pattern of silver-enhanced ZEN terminals in all these Timm modifications are too heterogeneous in quality, and even when most successful, i.e., when applied to rat brains, the staining is blurred and of little diagnostic or descriptive value. According to the present study, the reason is that the sulfidation takes place after the zinc ions have left their original location in the vesicles and most of them have been chemically bound to macromolecules and are no longer available for transformation into zinc sulfide crystal lattices.
When the vesicular zinc ion pool is chelated by diethyldithiocarbamate (DEDTC) in vivo (in rats) the present technique, like all other zinc-specific AMG techniques, results in blank AMG sections. This is because the zinc–DEDTC bonds cannot be broken by the sulfide ions, and it proves that the AMG pattern results from zinc ions. If the pattern were caused by copper ions the hypothetical copper–DEDTC would be transformed into copper sulfide crystals that could be AMG silver-enhanced (Danscher et al. 1985; Danscher and Montagnese 1994; Danscher 1996).
It can be concluded, therefore, that it is imperative that the zinc ion pool in the synaptic vesicles of ZEN neuronal terminals is chemically bound as stable crystals before it can escape from the vesicles. In animals this can be done in several ways: by transcardial perfusion (Danscher 1981); by in vivo exposure to sulfide ions (Danscher 1996); or by in vivo treatment with sodium selenite/selenide either intravenously, intraperitoneally, or intracerebrally (Danscher 1981,1982,1994). In tissue sections, the only way of making sure that the zinc ions are trapped and stay localized is to keep the tissue frozen until the chemical binding has taken place. It is therefore of the utmost importance that the biopsy material is frozen as soon as possible in liquid nitrogen or by CO2 gas and kept frozen until the zinc ions are captured by sulfide ions in the gas chamber. However, when tissue has been frozen it can be stored for an extended period of time with only minor leakage of zinc ions and, as a result, almost no decrease in the quality of the AMG staining.
AMG analysis of human brains 6 hr post mortem has given most unsatisfactory results because of the diffusion of zinc ions from the zinc ion-containing synaptic vesicles of ZEN boutons, a process that most likely starts as soon as the terminals are deprived of oxygen. Therefore, only biopsies from surgical or diagnostic intervention in the human brain can be expected to give an optimal AMG demonstration of the ZEN terminals. However, the possibility that human brain autopsy specimens taken shortly after death could still contain appreciable amounts of sulfide-available zinc ions in their ZEN terminals cannot be excluded. In rat, a usable AMG staining of the ZEN terminals can be obtained from animals that have died 2 hr before the brain was removed.
The “autoTimm” method shows some staining in, i.e., hippocampus and amygdala after 24 hr (Danscher and Zimmer 1978). This AMG staining of late postmortem brain areas known to have a particularly high level of ZEN terminals is caused by release and subsequent binding of sulfide ions to still free zinc ions. Because of the decayed tissue, this approach is of limited value.
The finding of an AMG–zinc pattern in biopsies from human neocortex that appears to be almost identical to the pattern of ZEN terminals in the rat brain makes the technique a promising tool for future studies in this particular population of neurons in the human brain, particularly since new studies suggest an increase in the zinc content of hippocampus and amygdala in Alzheimer-diseased brains (Constantinidis 1990; Deibel et al. 1996; Danscher et al. unpublished).
The presence of zinc ions in synaptic vesicles of human ZEN neurons suggests that ZEN neurons in humans and animals might be phylogenetically identical.
Apart from its use in brain studies and possible future use in pathological diagnostics, the present AMG technique can be applied to sections from many organs that contain zinc-enriched cells, e.g., Paneth cells in the gut, β-cells in the Langerhans islets of the pancreas, secretory cells in prostate and epididymis, or zinc ions in semen.
Footnotes
Acknowledgements
Supported by the Danish Medical Research Council, Direkt⊘r E. Danielsen og Hustrus Fond, Beckett-Fonden, and Karen Elise Jensens Fond.
We thank Prof H. Su for valuable comments on the manuscript and Ms H. Brandstrup, Ms D. Jensen, Mr A. Meier, Mr T.A. Nielsen, and Ms K. Wiedemann for expert technical assistance.