
Abstract
Neural stem cells (NSC) with self-renewal and multilineage potential are considered good candidates for cell replacement of damaged nervous tissue. In vitro experimental conditions can differentiate these cells into specific neuronal phenotypes. In the present study, we describe the combined effect of basic fibroblast growth factor (bFGF) and dibutyryladenosine 3',5'-cyclic monophosphate (dbcAMP) on the differentiation of fetal rat striatal NSC into tyrosine hydroxylase-positive cells. Tyrosine hydroxylase induction was accompanied by the activation of ERK1/ERK2 mitogen-activated protein kinase and was inhibited by the ERK1/ERK2 pathway blocker PD98059, suggesting that ERK activation may be important for this process. In addition, protein kinase C (PKC) was shown to be required for tyrosine hydroxylase protein expression. The inhibition of PKC by staurosporin, as well as its downregulation, decreased the ability of bFGF+dbcAMP to generate tyrosine hydroxylase-positive cells. Moreover, the PKC activator phorbol 12-myristate 13-acetate (PMA) together with bFGF and dbcAMP led to a significant increase in phospho-ERK1/ERK2 levels, and the percentage of β-tubulin III-positive cells that expressed tyrosine hydroxylase increased by 3.5-fold. PMA also promoted the phosphorylation of the cyclic AMP response element binding protein that might contribute to the increase in tyrosine hydroxylase-positive cells observed in bFGF+dbcAMP+PMA-treated cultures. From these results, we conclude that the manipulation in vitro of NSC from rat fetal striatum with bFGF, cyclic AMP analogs, and PKC activators promotes the generation of tyrosine hydroxylase-positive neurons.
N
Members of the FGF family and neurotrophins cooperate with agents that increase cAMP levels to induce TH expression in primary cultures of noncatecholaminergic neurons (Du and Iacovitti 1997a; Pliego Rivero et al. 1999) and in a cell line derived from human teratocarcinoma cells (Iacovitti and Stull 1997). Furthermore, protein kinase C (PKC) activators significantly increase TH production in these cells (Du and Iacovitti 1997b; Iacovitti et al. 2001). Neurons derived from mesencephalic human progenitors, which spontaneously express TH but low concentrations of dopamine, respond to brain-derived neurotrophic factor (BDNF) and cAMP with increased TH expression and dopamine secretion (Riaz et al. 2002). However, to our knowledge, the combined effect of trophic factors (such as bFGF or BDNF) and cAMP analogs to induce de novo TH expression in the progeny of NSC, which spontaneously do not generate dopaminergic neurons, has not been reported.
In the present study, we have analyzed some in vitro conditions for promoting the differentiation of EGF-expanded NSC derived from fetal rat striatum (striatal EGF-NSC) into TH-positive cells and the involvement of specific intracellular signaling pathways in this effect. Differentiated cultures derived from striatal EGF-NSC do not express TH spontaneously, but when exposed simultaneously to bFGF and dibutyryladenosine 3',5'-cyclic monophosphate (dbcAMP), TH expression is observed. The cooperation of bFGF and dbcAMP to induce TH depends on the developmental stage of the cultures. In fact, TH-positive cells are only observed when both factors are applied to cultures after differentiated neurons and glial cells are present. Our study shows that the activation of the ERK1/ERK2 mitogen-activated protein kinase (MAPK) signaling pathway is required for the generation of TH-positive cells in striatal EGF-NSC progeny. In addition, results from this study suggest that PKC also mediates TH protein expression in these cells. The inhibition of PKC by staurosporin or PKC downregulation significantly decreases the ability of bFGF and dbcAMP to induce TH. Moreover, the PKC activator phorbol 12-myristate 13-acetate (PMA) acts synergistically with bFGF and dbcAMP to significantly increase the number of TH-positive neurons by 3.5-fold. The role of nuclear ERK1/ERK2 MAPK and the transcription factor cAMP response element binding protein (CREB) in the induction of TH expression is also discussed.
Materials and Methods
Cell Cultures
Striatal primordia from E15 Sprague-Dawley rat embryos were dissected and mechanically dissociated. Cell suspensions were grown in a defined medium (DF12) composed of DMEM and Ham's F-12 (1:1), 2 mM
Differentiation of Striatal EGF-NSC
After a minimum of four passages, cells were plated at a density of 20,000–30,000 cells/cm2 on 15 μg/ml poly-
Immunocytochemical Staining
The polyclonal antibodies used in this study were anti-β-tubulin III (BabCO; Richmond, CA), phospho-CREB (Upstate Biotechnology; Lake Placid, NY), TH (Chemicon International; Temecula, CA), ERK1/ERK2 MAPK (Sigma), and GFAP (Dako; Glostrup, Denmark). Monoclonal antibodies against β-tubulin III and phospho-ERK1/ERK2 MAPK were obtained from Sigma, anti-TH was obtained from Chemicon, and anti-nestin (clone Rat 401) was from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IO). Monoclonal antibodies against A2B5, O4, and O1 were obtained in our laboratory as hybridoma supernatants. Secondary antibodies, goat anti-rabbit and sheep anti-mouse conjugated with rhodamine (1:100) and FITC (1:100), were purchased from Jackson Immunoresearch Laboratories, Inc. (West Grove, PA), and Boehringer Mannheim, respectively.
For immunocytochemical studies, cells were fixed with 4% paraformaldehyde for 10 min and immunostained for A2B5 (1:10), O4 (1:10), O1 (1:10), GFAP (1:500), TH (1:500), and β-tubulin III (1:200 for monoclonal and 1:3000 for polyclonal anti-β-tubulin III), as previously described (Bazán et al., 1998). Cover slips were mounted in a medium containing p-phenylenediamine and bisBenzimide (Hoechst 33342; Sigma).
Double labeling of phospho-CREB (1:100) and phospho-ERK1/ERK2 MAPK (1:100) was sequentially performed using an immunoperoxidase procedure for phospho-CREB detection in which the secondary anti-rabbit antibody was conjugated to peroxidase (1:50; Jackson). The phospho-ERK1/ERK2 MAPK antigen was visualized with a secondary anti-mouse antibody conjugated to alkaline phosphatase (1:100; Chemicon) and Fast Red TR/Naphthol AS-MX (Sigma).
Proliferation Studies
To enable the detection of proliferating cells, 50 μM bromo-deoxyuridine (BrdU), a marker of DNA synthesis, was added simultaneously with bFGF, dbcAMP, PMA or their combinations to 7-dpp cultures. Twenty-four to 48 hr later, cells were fixed, permeabilized with ethanol acetic solution (19:1), and treated with 2 N HCl for 30 min at 4C. Primary monoclonal antibody against BrdU (1:20; Dako) was added for 1 hr at room temperature. BrdU incorporation was detected as previously described (Reimers et al. 2001).
Terminal Deoxynucleotidyl Transferase-mediated Biotinylated UTP Nick End Labeling Staining
DNA fragmentation was evaluated using the terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated UTP nick end labeling (TUNEL) technique. Briefly, cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate buffer (pH 7.4) for 2 min at 4C, and incubated with 5 units of TdT and 10 mM dUTP-biotin (Boehringer Mannheim) in Tris buffer saline (pH 7.6) for 1 hr at 37C. TUNEL detection was performed using a biotin-linked secondary antibody (1:200; Vector Laboratories, Inc., Burlingame, CA) followed by incubation with an avidin-biotinylated horseradish peroxidase complex (Vectastain Elite ABC Kit; Vector). Finally, 0.05% 3,3-diaminobenzidine in 0.05% H2O2 and 0.6% NiCl was used as chromogen.
Phosphoinositide Turnover
Seven-dpp cultures labeled for 24 hr with 1.5 μCi/ml [3H]-inositol (21 Ci/mmol; New England Nuclear, Dreiech, West Germany) were stimulated for 15 min with 10 ng/ml bFGF. The reaction was stopped by adding 1 ml of cold methyl alcohol and 0.12 M HCl (1:1), and fractions containing inositol-monophosphate were determined by ion-exchange chromatography, as previously described (Bazán et al. 1998).
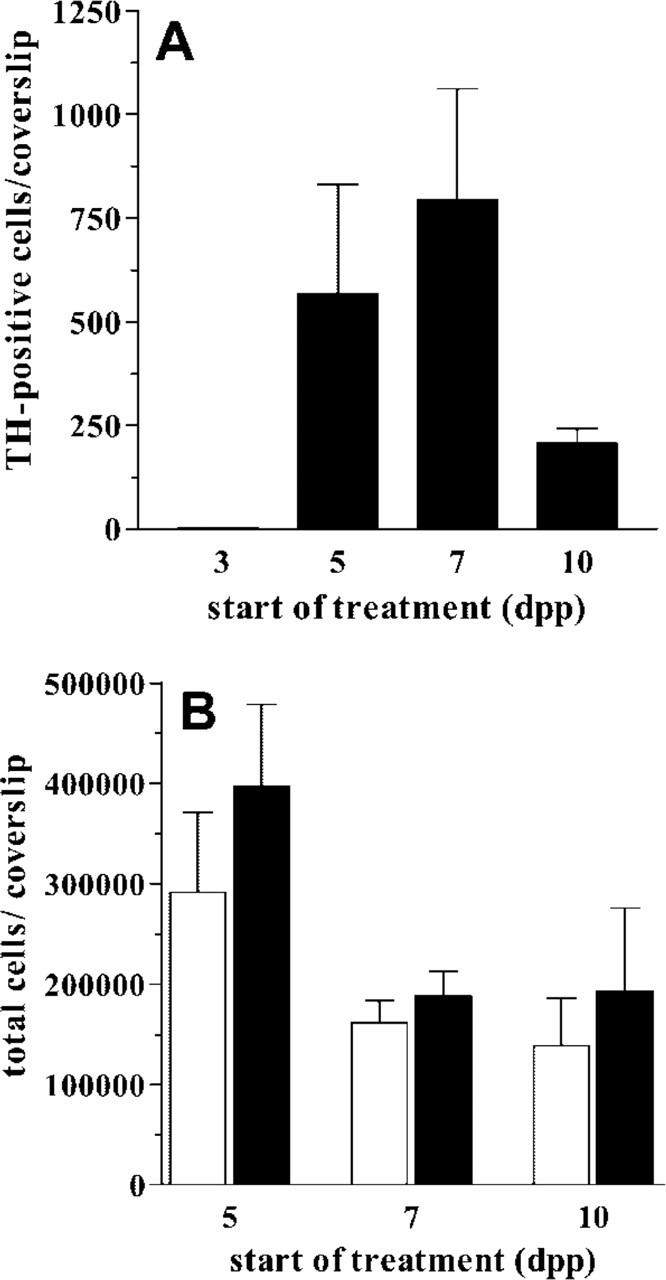
TH induction by bFGF + dbcAMP treatment during the in vitro differentiation of striatal EGF-NSC. At 5, 7, or 10 dpp, a 24-hr treatment with 10 ng/ml bFGF and 1 mM dbcAMP (bFGF + dbcAMP) induces TH expression in striatal EGF-NSC (
Western Blot Protein Analysis of Phospho-ERK1/ERK2 and Phospho-CREB
NSC progeny were treated for 30 min with TH inducers, and proteins were processed for Western blot analysis to determine the relative levels of phospho-ERK1/ERK2 and phospho-CREB. Cells were lysed with 0.5 M Tris-HCl buffer (pH 7.4) containing 0.24% Triton X-100, 10 μg/ml leupeptin, and 0.5 mM PMSF, all from Sigma. After 1 hr at 4C, samples were centrifuged at 12,000 × g for 30 min. Total protein content was quantified using a BCA kit (Pierce; Rockford, IL). Aliquots of 40 μg of protein were separated by electrophoresis on 10% SDS-polyacrylamide minigels and transferred to nitrocellulose filters. Membranes were soaked in blocking solution (0.2 M Tris-HCl, 137 mM NaCl, and 3–5% dry skimmed milk, pH 7.6) and incubated with primary antibodies diluted in the same blocking solution: anti-phospho-ERK1/ERK2 MAPK (1:5000), anti-ERK1/ERK2 MAPK (1:10,000), and anti-phospho-CREB (1:1000). After extensive washing, membranes were incubated with the peroxidase-conjugated secondary antibodies diluted 1:1000 in blocking solution. The filters were developed with enhanced chemiluminescence Western blotting analysis, following the procedure described by the manufacturer (Amersham, Buckinghamshire, England). Membranes were immunolabeled for control charge using anti-ERK1/ERK2 MAPK antibodies. Autoradiograms were quantified by computer-assisted videodensitometry.
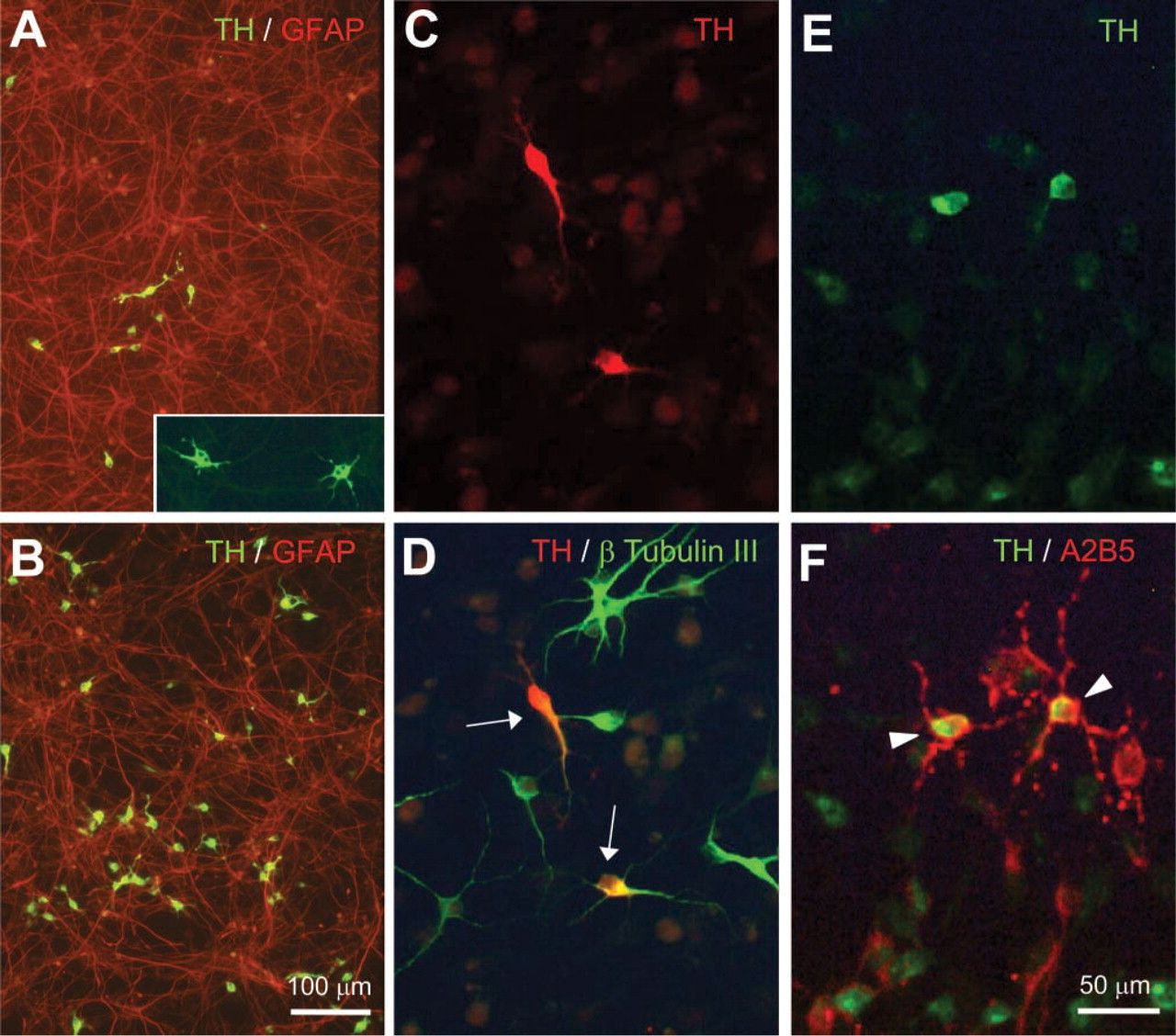
TH expression in striatal EGF-NSC progeny. At 7 dpp, cultures were treated for 24 hr with 10 ng/ml bFGF plus 1 mM dbcAMP (
Data Analysis and Cell Counting
Results are expressed as means ± SEM from three to eight independent experiments done in triplicate or quadruplicate. Where indicated, data represent means ± SEM of several cover slips. For each cover slip, stereological sampling of 25 visual fields (magnification of 200× or 400×) was performed by fluorescence microscopy. The number of cells was corrected for cover slip area. Statistical analyses were performed using Student's t-test or one-way ANOVA followed by Bonferroni's multiple comparison test, and differences were considered significant at p≤0.05.
Results
TH Induction by bFGF and dbcAMP Treatment during Striatal EGF-NSC Differentiation
Striatal EGF-NSC were plated onto adherent substrate in the presence of EGF for 3 days to enhance the expansion of precursor cells. After this period, EGF was withdrawn and cells were grown in defined medium that promotes their differentiation into neurons, oligodendrocytes, and astrocytes following the specific temporal pattern that has been described elsewhere (Bazán et al. 1998; Reimers et al. 2001). Neurons generated from striatal EGF-NSC did not express TH at 3, 5, 7, and 10 dpp. Twenty-four hr of treatment with 10 ng/ml bFGF or 1 mM dbcAMP alone did not induce TH expression at any experimental time. However, the combination of 10 ng/ml bFGF plus 1 mM dbcAMP (bFGF+dbcAMP) promoted TH protein expression in some cells from striatal EGF-NSC progeny. This treatment was effective when applied to 5, 7, or 10-dpp cultures but not at earlier stages of differentiation (Figure 1A). Moreover, this treatment did not change the total number of cells with respect to untreated cultures (Figure 1B).
TH-positive cells generated from striatal EGF-NSC showed a protoplasmic morphology with short neuritic extensions and polymorphic, often bilobulated nuclei, suggesting an immature neuronal phenotype (Figure 2A, inset). Most TH-positive cells (93 ± 2.2%, n=3) expressed the neuronal marker β-tubulin III (Figures 2C and 2D) and represented 3.5 ± 0.7% of the total neuronal population. Although 16–20% of them were positive for A2B5 (Figures 2E and 2F), none of the TH-positive cells colabeled with GFAP (Figure 2A), O4, or O1. These results indicate that the glial progeny derived from striatal EGF-NSC did not express TH after bFGF + dbcAMP treatment.
The individual actions of bFGF and dbcAMP on TH induction were dose dependent. Using a fixed concentration of dbcAMP (1 mM), the maximal effect of bFGF on the number of TH-positive cells was achieved between 10 and 50 ng/ml (Figure 3A). Similarly, at a fixed concentration of bFGF (10 ng/ml), dbcAMP action was maximal at concentrations of 1–2 mM (Figure 3B). TH induction in striatal EGF-NSC progeny was also time dependent. In the presence of bFGF+dbcAMP, TH immunoreactivity was observed at 6 hr after treatment and peaked 12–24 hr later (Figure 4A). This time-dependent increase in the number of TH-positive cells was not attributable to the proliferation of neural precursors because BrdU incorporation was unchanged after 24 hr of bFGF+dbcAMP treatment [2.45 ± 0.35% (n = 10) and 2.08 ± 0.08% (n = 9) BrdU-positive cells in untreated and bFGF+ dbcAMP-treated cultures, respectively]. Moreover, none of the TH immunoreactive cells incorporated BrdU (data not shown).
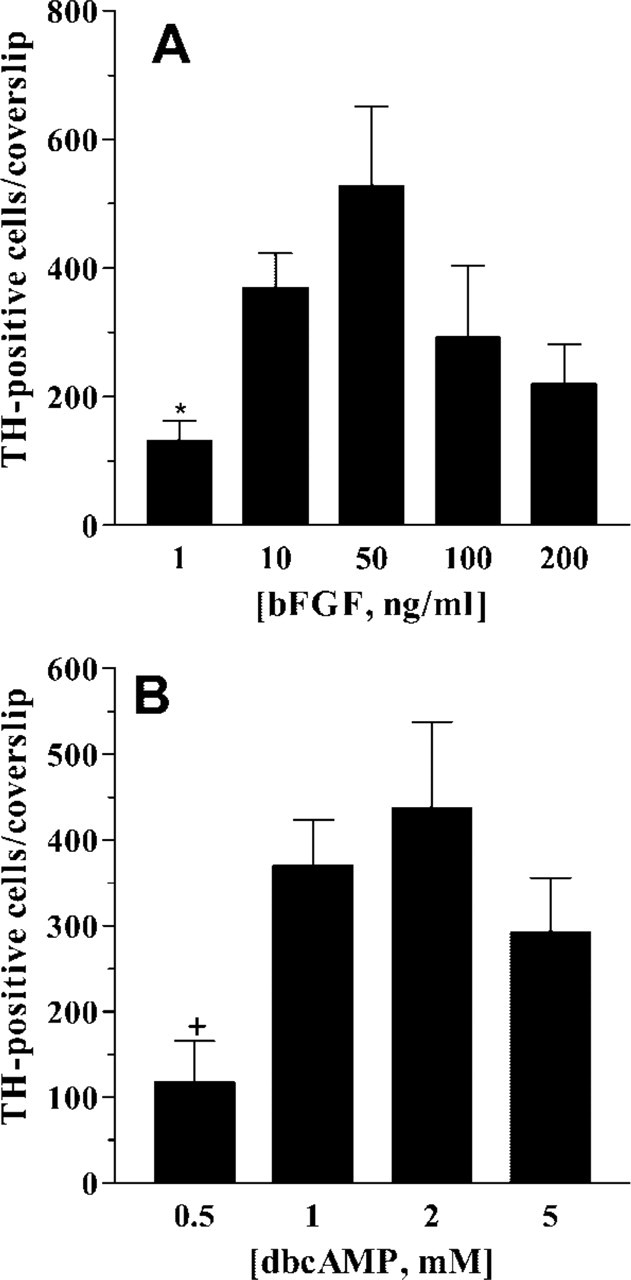
Dose-response curves for bFGF and dbcAMP on TH induction. At 7 dpp, cultures were treated for 24 hr with 1 mM dbcAMP in combination with increasing doses of bFGF. Under these experimental conditions, maximal TH induction was achieved with 10–50 ng/ml bFGF (
As shown in Figure 4A, the number of TH-positive cells decreased from 48 to 72 hr of treatment, whereas the total number of β-tubulin III-positive cells remained unchanged at all experimental times studied (Figure 4B). From these results, we conclude that the loss of TH-positive cells is not directly related to the possible loss in the number of neurons. On the other hand, TUNEL analysis showed a slight but significant decrease in the number of TUNEL-positive cells after 48 hr of bFGF+dbcAMP treatment (Figure 4C).
Other factors, such as some members of the FGF family (acidic FGF [aFGF] and FGF8), BDNF, and glial-derived neurotrophic factor (GDNF), alone or in combination with dbcAMP, were tested for their ability to induce the expression of TH. Only the combination of aFGF (10 ng/ml) plus dbcAMP (1 mM) promoted the generation of TH-positive cells in a small proportion of β-tubulin III-positive cells (0.697 ± 0.115%, n=3).
Besides TH induction, bFGF+dbcAMP treatment also affected the morphology of GFAP-positive cells, which changed from a stellate aspect with multiple and short extensions (Figure 5A) to a fibrillar morphology with long processes (Figure 5B). Similar results were found in aFGF+dbcAMP-treated cultures, whereas FGF8 + dbcAMP treatment did not promote any visible change in the morphology of GFAP-positive cells (data not shown).
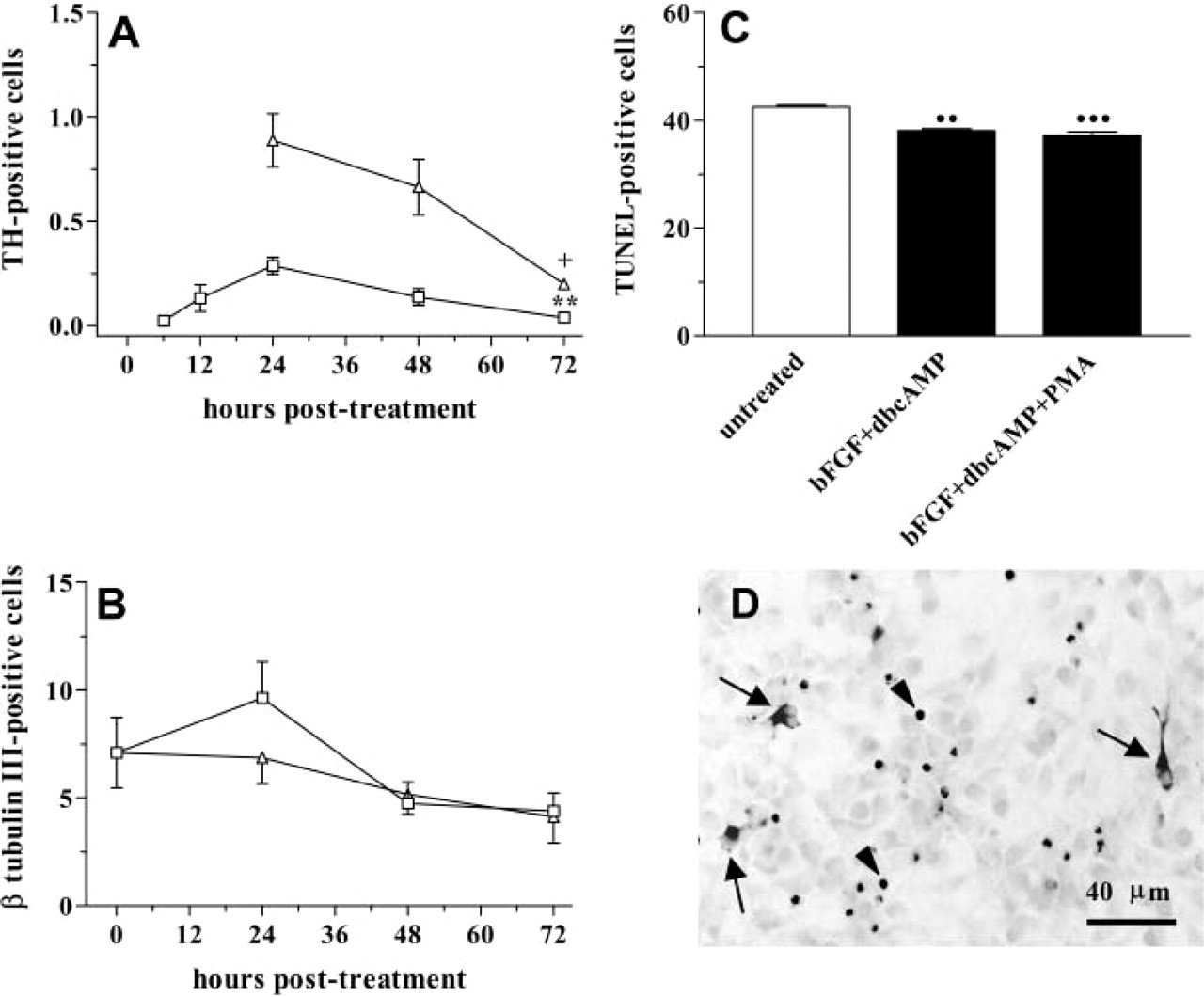
Survival period of in vitro-generated TH-positive cells. Almost two thirds of TH-positive cells generated with 10 ng/ml bFGF plus 1 mM dbcAMP (
Roles of ERK1/ERK2 MAPK, PKC, and CREB in TH Induction
By using a monoclonal antibody that specifically recognizes the active (phosphorylated) form of ERK1 and ERK2 MAPK (44 and 42 kD, respectively), we tested the ability of bFGF and/or dbcAMP to activate these MAPKs in striatal EGF-NSC progeny. Western blot analysis revealed no detectable levels of phospho-ERK1/ERK2 in untreated or 1 mM dbcAMP-treated cultures. By contrast, 7-dpp cultures treated for 30 min with 10 ng/ml bFGF showed a significant increase in phospho-ERK1/ERK2 levels, with no change in the total amount of ERK1/ERK2 (Figure 6A). Similar results were obtained with bFGF+dbcAMP treatment, but the phosphorylation of ERK1/ERK2 was greater (Figure 6A). To study if activated MAPK was involved in de novo TH expression, cultures were treated with bFGF+dbcAMP in the presence of 10−-7 to 2 × 10−-5 M PD98059, a specific inhibitor of MAPK as well as its upstream kinase (MEK). Twenty-four hr later, TH immunoreactivity was analyzed. As shown in Figure 6B, PD98059 inhibited the ability of bFGF+dbcAMP treatment to induce TH protein expression in a dose-dependent manner (IC50 = 2.19 ± 0.76 μM, n=3). However, the total number of cells and the morphological changes induced by this treatment in the glial progeny were not affected by PD98059.
The binding of bFGF to FGF receptors (FGFRs) could lead to the activation of phospholipase C7 (PLC7), which promotes the release of Ca2+ from internal stores and, combined with the generation of diacylglycerol, activates PKC. At 7 dpp, treatment with 10 ng/ml bFGF for 15 min increased the levels of inositol monophosphate by 2.15 ± 0.067-fold (n = 3), as demonstrated by ion-exchange chromatography. These results suggest that the PLC7-PKC signaling pathway is activated by FGFRs in striatal EGF-NSC. Staurosporin, a relatively specific inhibitor of PKC, did not affect cell culture viability but inhibited both bFGF+ dbcAMP-induced TH expression (Figure 7A) and the morphological changes in GFAP-positive cells promoted by this treatment (Figure 5C). Moreover, the downregulation of PKC mediated by sustained activation with 10−-7 M PMA before the addition of bFGF+dbcAMP also decreased the ability to generate TH-positive cells (Figure 7A). These results strongly suggest that PKC activation is needed for TH induction by bFGF+dbcAMP.
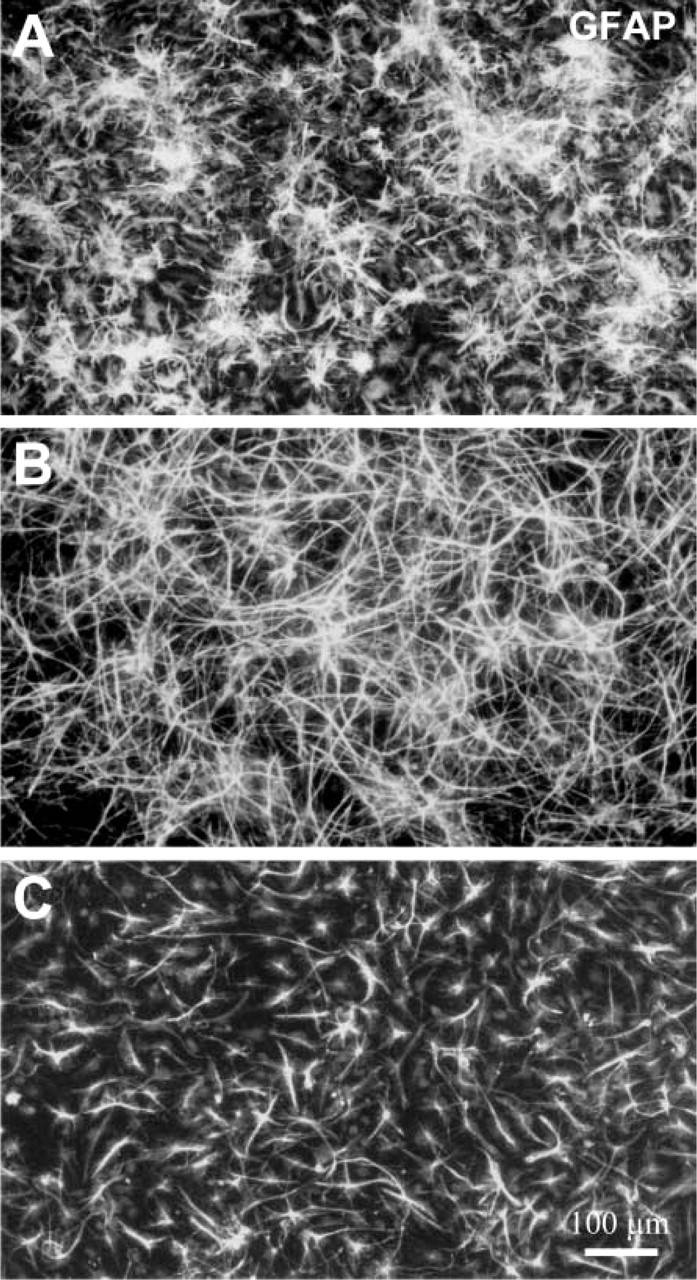
Morphological changes in astroglia under TH-inductive conditions. At 7 dpp, cultures of striatal EGF-NSC were treated with 10 ng/ml bFGF plus 1 mM dbcAMP, and GFAP immunostaining was analyzed 24 hr later. The morphology of GFAP-positive cells changed from stellate with multiple and short extensions (
On the other hand, we observed that the PKC activator PMA was a weak TH inducer (≃60 TH-positive cells per cover slip). The combination of 10−-7 M PMA and 1 mM dbcAMP (dbcAMP+PMA) was as effective as bFGF + dbcAMP at inducing TH expression (Figure 7B). The maximal number of TH-positive neurons was found when 10−-7 M PMA was applied for 24 hr in the presence of 10 ng/ml bFGF (bFGF+PMA) or 10 ng/ml bFGF plus 1 mM dbcAMP (bFGF+ dbcAMP+PMA) (Figures 2B and 7B). The number of TH-positive cell decreased after 48–72 hr of bFGF+ dbcAMP+PMA treatment without affecting the total number of β-tubulin III-positive cells (Figures 4A and 4B). The number of TUNEL-positive cells also decreased in bFGF+dbcAMP+PMA-treated cultures (Figure 4C). Moreover, features of apoptotic nuclei in TH-positive cells were not observed at any of the post-treatment times analyzed (Figure 4D).
As shown in Figure 6A, 30 min of treatment with bFGF+dbcAMP+PMA produced a significant increase in phospho-ERK1/ERK2 levels. Immunocytochemical studies showed a cytoplasmic and nuclear localization of phospho-ERK1/ERK2 in cultures treated with bFGF+ dbcAMP+PMA (Figure 6F, inset), whereas those cultures treated with bFGF+dbcAMP only showed the cytoplasmic localization of these MAPKs (Figure 6E, inset). The nuclear and cytoplasmic staining of phospho-ERK1/ERK2 was also observed after 3 hr of treatment but not 6 hr later (data not shown). When the ERK1/ERK2 pathway blocker PD98059 was applied to the cultures 15 min before bFGF+dbcAMP+ PMA treatment, the number of TH-positive cells was significantly reduced with respect to those cultures treated with bFGF+dbcAMP+PMA (Figure 6C).
CREB is the main transcription factor that binds to the cAMP response element, a well-characterized element within the rat TH gene promoter (Kumer and Vrana 1996). Because CREB may be activated by different stimuli, including trophic factors and protein kinase activators, it seemed reasonable to test its possible role in the de novo induction of TH in striatal EGF-NSC. Western blot analysis revealed that 30 min of treatment with 1 mM dbcAMP produced a slight but consistent increase of phospho-CREB levels in striatal EGF-NSC progeny compared with untreated cultures (Figure 6A). However, no significant changes were observed in other experimental situations. By contrast, immunocytochemical analysis showed that 30 min to 3 hr of treatment with bFGF+dbcAMP + PMA increased phospho-CREB nuclear staining in ≃17% of the cells (compare Figure 6D, untreated culture, with Figure 6F, bFGF + dbcAMP+PMA-treated cultures).
Discussion
The present study shows for the first time the combined effect of bFGF and dbcAMP to induce TH protein expression in striatal EGF-NSC progeny. This induction was dependent on the developmental stage of the cultures and was enhanced by the PKC activator PMA. Our results also suggest the relevance of ERK1/ERK2 activation and the possible involvement of the transcription factor CREB in the signaling pathway that led to TH induction.
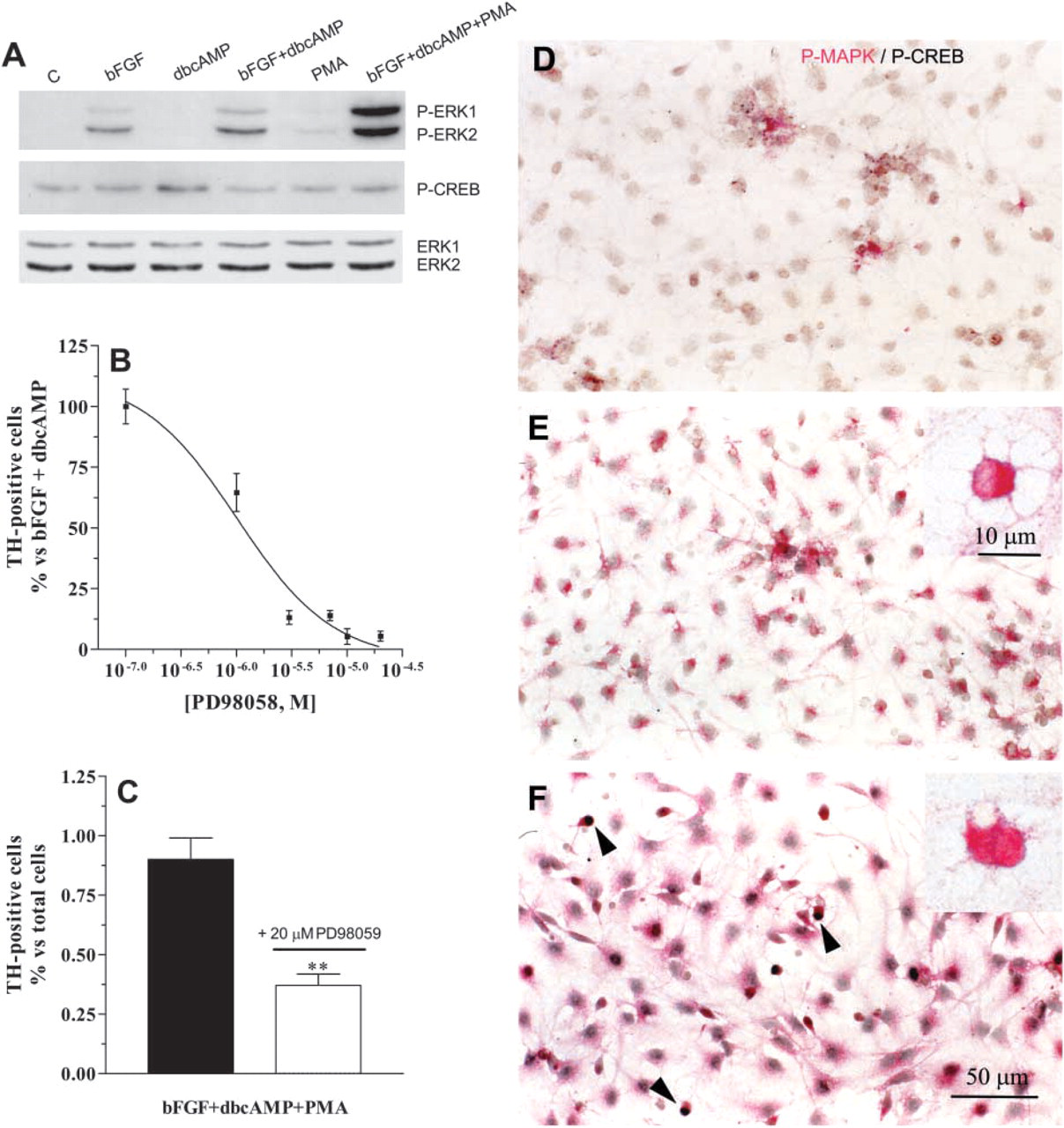
Role of ERK1/ERK2 MAPK and CREB on TH induction. At 7 dpp, cultures were treated for 30 min with vehicle [control (C)], 10 ng/ml bFGF, 1 mM dbcAMP, 10 ng/ml bFGF plus 1 mM dbcAMP, 10−-7 M PMA, or 10 ng/ml bFGF plus 1 mM dbcAMP plus 10−-7 M PMA. Immunoblot analyses of these cultures for the detection of phospo-ERK1/ERK2, phospho-CREB, and ERK1/ERK2 were performed (
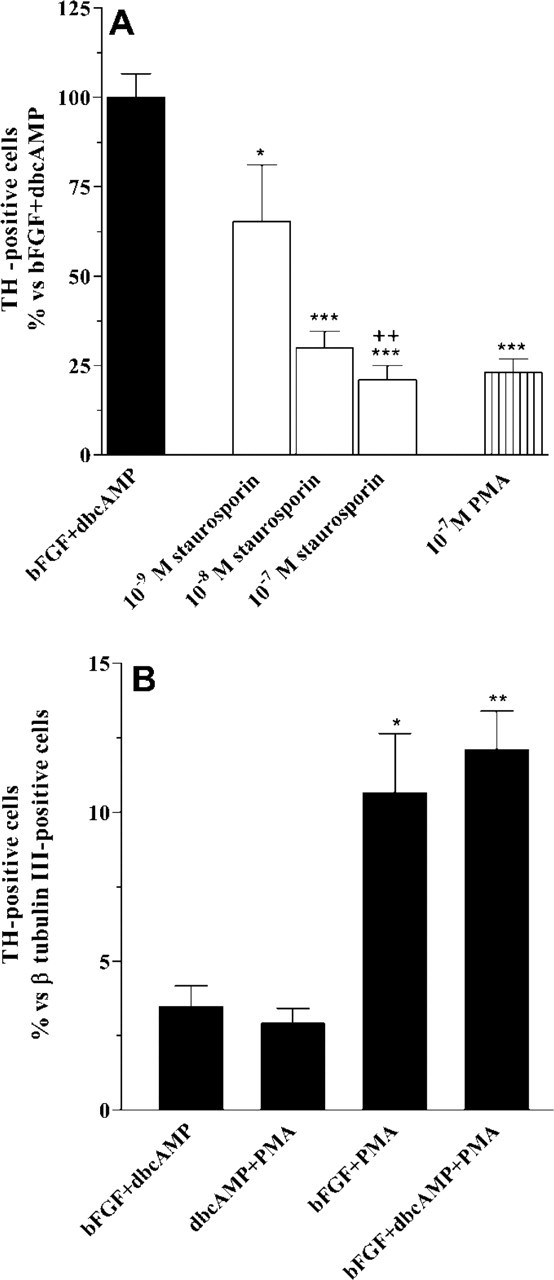
Role of PKC on TH induction. At 7 dpp, cultures of striatal EGF-NSC were treated for 30 min with 10−-9 to 10−-7 M staurosporin before the addition of 10 ng/ml bFGF plus 1 mM dbcAMP. Staurosporin inhibited the ability of bFGF + dbcAMP to induce TH in a dose-dependent manner (
Previous reports by Iacovitti's group have shown that aFGF and, to a lesser extent, bFGF require cooperation with protein kinase A (PKA) and PKC activators to induce TH protein expression in primary neuron cultures derived from rat embryonic striatum (Du and Iacovitti 1997a,b; Stull et al. 2001) and from human embryonic carcinoma (Iacovitti and Stull 1997; Iacovitti et al. 2001; Stull and Iacovitti 2001). We show here that the induction of TH by cooperation between bFGF and dbcAMP in striatal EGF-NSC is highly dependent on the developmental stage of the cultures. In fact, bFGF+dbcAMP treatment was not effective when applied at 3 dpp. Before 3 dpp, more than 70% of the cells were proliferating and only a small percentage expressed specific markers for neurons and glial cells (Reimers et al. 2001; Lobo et al. 2003). Other studies have reported that an EGF-propagated stem cell line failed to express TH after treatment with trophic factors and coactivators (Schinstine and Iacovitti 1997) and proposed that failure to obtain TH-positive cells was attributable to the developmental stage of the stem cells. Our results confirm this hypothesis, demonstrating the induction of TH-positive cells when bFGF and dbcAMP are applied at 5, 7, or 10 dpp. At these times, more differentiated neurons and glial cells are already present in the cultures and cell proliferation significantly decreases (Bazán et al. 1998; Reimers et al. 2001).
Although bFGF has mitogenic actions in NSC and their progeny (Daadi and Weiss 1999; Reimers et al. 2001), our BrdU pulse-labeling analyses showed no increase in BrdU incorporation under bFGF+dbcAMP treatment, and none of the TH-positive cells showed BrdU immunostaining. These results indicate that mitotically active neural precursors are not the target for TH induction. Most of the TH immunoreactive cells expressed the neuronal marker β-tubulin III, suggesting that postmitotic neurons derived from striatal EGF-NSC are the final targets for bFGF+dbcAMP inductive activity. A relatively small amount of TH-positive cells colabeled with A2B5, an antibody that recognizes bipotential O2A glial progenitors (Raff et al. 1984) and a subset of neurons (Schnitzer and Schachner 1982). In our cultures, 5% of A2B5-positive cells were also immunoreactive for β-tubulin III and showed a neuronal morphology (our unpublished observations). Because A2B5 labeling has been associated with cells exiting from the cell cycle (Przyborski et al. 2000), β-tubulin III/A2B5-positive cells could represent a population of newborn neurons that are able to respond to TH-inducing cues.
As mentioned above, aFGF has been reported to be more effective than bFGF to induce TH in different types of noncatecholaminergic neurons (Du and Iacovitti 1997a; Iacovitti and Stull 1997). However, in our hands, aFGF+dbcAMP treatment yielded a lower number of TH-positive cells in striatal EGF-NSC progeny than bFGF+dbcAMP. The absence of heparin in our media might explain this lower effectiveness. Although heparin is needed to increase the affinity of aFGF for its receptors, its use in our cultures affects cell adhesion to the substrate. Another member of the FGF family, FGF8, has been reported to increase the generation of dopaminergic neurons derived from mouse embryonic stem cells (Lee et al. 2000) but failed to induce TH expression in our striatal EGF-NSC cultures. This trophic factor shows high affinity for FGFR3 (Chellaiah et al. 1999), a FGFR that was found to be critical for the induction of TH expression in dopaminergic neurons in vivo (Ye et al. 1998). Although FGFR3 is expressed in neurons and glial cells derived from striatal EGF-NSC (Reimers et al. 2001), the low levels of FGFR3 protein observed in these cells (Lobo et al. 2003) could explain the lack of action of FGF8. Moreover, two other growth factors, BDNF and GDNF, which have been involved in the differentiation of mesencephalic progenitor cells into dopaminergic neurons (Ling et al. 1998; Storch et al. 2001; Riaz et al. 2002), were not able to induce TH protein expression in the striatal EGF-NSC progeny examined here. It should be considered that stem/progenitor cells probably represent heterogeneous cell populations in relation to their response to environmental cues.
Besides TH induction, bFGF+dbcAMP or aFGF+ dbcAMP treatment promoted morphological changes in GFAP-positive cells. Moreover, these treatments induced nestin reexpression and increased FGFR1 immunostaining (our unpublished observations). We have reported similar changes in striatal EGF-NSC treated with bFGF that were associated with the acquisition of a phenotype characteristic of reactive glia (Reimers et al. 2001). Reactive astrocytes might synthesize and release unidentified glial factors other than GDNF that, combined with dbcAMP, could promote the expression of TH in target cells. The idea that glial cells might mediate TH induction is supported by previous studies reporting the need of factors released by glial cells to induce the catecholaminergic phenotype in neural precursors (Daadi and Weiss 1999; Wagner et al. 1999).
Trophic factors and cAMP activate the ras/raf/MEK/ERK pathway in different cell types (Frodin et al. 1994; Vossler et al. 1997; Suzuki et al. 1999; Abe and Saito 2000). This signaling pathway has been proposed as a mediator of TH induction in noncatecholaminergic neurons (Guo et al. 1998; Stull and Iacovitti 2001; Stull et al. 2001). The present results indicate that the activation of ERK1/ERK2 participates in TH induction in striatal EGF-NSC. In fact, bFGF+ dbcAMP treatment promoted their phosphorylation, and PD98059, a specific inhibitor of ERK1/ERK2 phosphorylation, decreased the number of TH-positive neurons in a dose-dependent manner. It is important to point out that bFGF significantly increases phospho-ERK1/ERK2 levels but it does not induce TH expression. A possible explanation for the lack of action of bFGF is that there is a threshold for ERK1/ERK2 phosphorylation to lead to the promotion of TH protein expression. In striatal EGF-NSC, dbcAMP does not promote the phosphorylation of ERK1/ERK2, as has been reported in cultured astrocytes (Kurino et al. 1996). However, dbcAMP cooperates with bFGF to increase phospho-ERK1/ERK2 levels above those observed in bFGF-treated cultures. The synergistic activation of ERK1/ERK2 induced by co-stimulation of cAMP and growth factors has been observed in PC12 cells (Frodin et al. 1994), myeloid progenitors (Lee 1999), and striatal neurons (Guo et al. 1998). In striatal neurons, these changes in the level of phospho-ERK1/ERK2 were critical to induce increased binding of transcription factors from the Fos-Jun complex at the activator protein-1 regulatory element on the TH gene.
Several reports have related the activation of PKC to TH induction (Kedzierski et al. 1994; Du and Iacovitti 1997b; Najimi et al. 2002). Results from the present study strongly suggest the participation of PKC in TH expression: (a) the accumulation of inositol monophosphate on bFGF-treated cultures and results from other authors (Oury et al. 1992; Cross et al. 2002) indicate that bFGF can activate the PLC7-PKC pathway through the stimulation of FGFRs; (b) staurosporin partially blocks changes in the morphology of the astrocytes promoted by bFGF+dbcAMP treatment that, as mentioned above, might be necessary for TH induction; and (c) the addition of staurosporin to the cultures inhibits the effects of bFGF+dbcAMP on TH protein expression in a dose-dependent manner. Although staurosporin might inhibit other protein kinases differently than PKC, the fact that PKC down-regulation also reduces the ability of bFGF+dbcAMP to generate TH-positive cells strongly supports our hypothesis about the participation of PKC in TH induction.
On the other hand, the full activation of PKC by PMA in the presence of bFGF+dbcAMP yields a significant increase in the number of TH-positive neurons. The present combination of bFGF, dbcAMP, and PMA has proven to be very effective in the generation of TH-expressing cells in cultures derived from human cortex NSC (Paino et al. 2003). Like the present results, those experiments showed that there was a narrow time window after neural stem seeding on poly-
In different cell types, PMA actions are mediated through the activation of the raf/MEK/ERK pathway (Qiu et al. 2001; Chen and Davis 2003; Park et al. 2003). In the present study, PMA acts synergistically with bFGF + dbcAMP to promote a significant increase in the phosphorylation of ERK1/ERK2 that is associated with the increase in the number of TH-positive neurons observed in bFGF+dbcAMP+PMA-treated cultures. In fact, in the presence of 20 μM PD98059, the number of cells induced to express TH was decreased to less than one half. Similar results have been found in primary cultures of striatal neurons, in which the application of PMA to a cocktail containing aFGF, forskolin, and catecholamines increased the phosphorylation of ERK1/ERK2 and the number of TH-positive neurons (Guo et al. 1998).
In striatal EGF-NSC progeny, PMA also promotes the nuclear localization of phospho-ERK1/ERK2 where they might activate different transcription factors (Karin and Hunter 1995). The transcription factor CREB has been proposed as a major component in the regulation of TH gene expression by trophic factors and cAMP (Theofilopoulos et al. 2001; Riaz et al. 2002). Our Western blot analyses did not show significant changes in phospho-CREB levels under bFGF+dbcAMP or bFGF+dbcAMP+PMA experimental conditions. However, cultures stimulated with bFGF+dbcAMP+PMA showed an increase in phospho-CREB-positive cells. Because the increase in phospho-CREB nuclear staining was observed in 17% of cells, these small changes might not be detected by immunoblot techniques. Conceivably, phospho-ERK1/ERK2 mediate the activation of CREB, and the most likely molecule coupling ERK1/ERK2 to CREB phosphorylation is RSK2, a kinase that is activated by MAPK and phosphorylates CREB at serine-133 (Xing et al. 1996; Roberson et al. 1999). In addition, CREB phosphorylation could be directly elicited by PKC, because phospho-CREB-positive cells were also observed in PMA-treated cultures (our unpublished observations), an experimental condition in which phospho-ERK1/ERK2 levels were very low and a small population of TH-positive cells were found. Another possibility is the activation of the cAMP/PKA pathway by phorbol esters, as has been reported in smooth muscle cells (Baillie et al. 2001) and PC12 cells (Piech-Dumas et al. 2001). The activation of PKA by PMA might contribute to the phosphorylation of CREB and could explain why bFGF + PMA treatment was almost as effective as bFGF+dbcAMP+PMA for TH induction. Further experiments are warranted to determine if this signaling pathway mediates PMA actions.
Finally, the present data show that TH-positive neurons generated from striatal EGF-NSC significantly disappeared a few days after their induction. During this period, the number of neurons remained unchanged and the percentage of TUNEL-positive cells was decreased. Furthermore, no TH-positive/TUNEL-positive cells were elicited by TH-inductive treatments. These results indicate that loss of TH-positive neurons probably is not attributable to apoptotic cell death of neurons. However, we cannot exclude a loss of TH-positive cells through necrotic cell death or a quick removal of the apoptotic cells because striatal EGF-NSC show an important phagocytic activity (Lobo et al. 2003). Alternatively, bFGF and protein kinase activators could turn on TH gene expression but may be unable to sustain it, as has been proposed for TH induction by bone morphogenetic proteins in striatal neurons (Stull et al. 2001). The maintenance of TH expression may depend on signals or trophic support provided by other cells not present in the cultures. The addition of several factors (BDNF, GDNF, ascorbic acid, bFGF, dbcAMP) involved in the survival of dopaminergic neurons could not hold up TH expression for longer periods of time in culture (our unpublished observations). However, preliminary data from our laboratory indicate that bFGF+ dbcAMP+PMA in vitro-generated TH-positive cells survive for a minimum of 7 days after their transplantation into the adult striatum. These results suggest that the adult brain contains the appropriate factors or cells needed for the survival and/or maintenance of TH-positive cells generated from striatal EGF-NSC.
In summary, bFGF and dbcAMP induce de novo TH expression in postmitotic neurons derived from striatal EGF-NSC. This induction could be mediated by ERK1/ERK2 and PKC signaling pathways, whose pharmacological manipulation might be a useful strategy for stem cell-based replacement therapies.
Footnotes
Acknowledgements
This work was funded by the Fondo de Investigaciones Sanitarias (FIS 97/269) and Comunidad Autónoma de Madrid (CAM 8.1/4.5/99). CR and MVTL were recipients of FIS and CAM fellowships, respectively.
We are grateful to Dr M.L. Shelanski (Columbia-Presbyterian Medical Center, New York, NY) for comments and critical reading of the manuscript. We thank M.J. Asensio for technical help.