
Abstract
Leishmania amazonensis, an obligatory intracellular parasite, survives internalization by macrophages, but no information is available on the involvement of microglia. We have investigated microglia-protozoa interactions in mixed glial cultures infected with promastigote forms of L. amazonensis after lipopolysaccharide (LPS) or dexamethasone (DM) treatment. After 2 hr of exposure to parasites in control cultures, there was a small number of infected microglia (1%). Preincubation with LPS or DM led to 14% or 60% of microglial cells with attached parasites, respectively. DM treatment resulted in 39% of microglial cells with internalized parasites (controls or LPS-treated cells had ≤1%). Scanning electron micrographs showed numerous filopodia in DM-treated cells, whereas these projections were rarely observed in LPS-treated or control cells. DM treatment also affected the intramicroglial survival of Leishmania. In control cultures, internalized parasites, tagged with an anti-lipophosphoglycan (anti-LPG) antibody, showed fragmented DNA [terminal deoxyribonucleotide transferase-mediated dUTP-X nick end labeling (TUNEL+)] after 4 hr of interaction, but changes seemed slightly delayed in DM-treated cultures. After 12 hr, there were no LPG+/TUNEL+ profiles in controls, whereas rare LPG+ profiles still persisted in DM-treated cells. Our results suggest that microglia are highly effective in the elimination of Leishmania and that the process can be effectively studied by LPG/TUNEL double labeling.
M
Leishmania are obligatory intracellular pathogenic parasites that must gain entrance into mononuclear phagocytes to successfully complete their cell cycle and parasitize various species of mammalian hosts (for reviews, see Cunningham 2002; Sacks and Sher 2002; Vannier-Santos et al. 2002). However, it is not known how the “macrophages” of the central nervous system (the microglia) interact with this obligatory intracellular parasite in their physiological environment, i.e., in the presence of other major components of the neural tissue, such as astrocytes. Thus, we have chosen to study the interactions of Leishmania amazonensis with microglia in mixed glial cultures from neonatal tissue and compare these interactions with known features of Leishmania–macrophage interactions. We have also used treatment of microglia in mixed glial cultures with immune modulators [lipopolysaccharide (LPS) or dexamethasone (DM)] before interaction with L. amazonensis and have followed the destiny of the parasites by their specific lipophosphoglycan (LPG) membrane labeling plus terminal deoxyribonucleotide transferase-mediated dUTP-X nick end labeling (TUNEL) staining.
We have found that, in contrast to macrophages (Kane and Mosser 2000; for reviews, see Cunningham 2002; Vannier-Santos et al. 2002), microglia eliminate intracellular parasites very rapidly and that this process seems to be slowed by DM treatment. Our results indicate that microglia represent a highly effective barrier to the invasion of the brain by L. amazonensis and suggest that an extensive investigation of the mechanisms involved may provide additional clues to microglial physiological functions vis-a-vis intracellular parasites.
Materials and Methods
Primary Glial Cell Cultures
Forebrains isolated from newborn Swiss mice were dissected out, minced, and incubated at 37C with 0.002% trypsin (Difco; Detroit, MI) in Ca2+/Mg2+-free 0.85% PBS for 5 min. The brain cells were dissociated by successive cycles of mild incubation with trypsin, with periodic interruption of the enzymatic activity by the addition of 10% fetal calf serum (FCS) (Sigma Chemical; St. Louis, MO). After tryp-sinization, the cell suspension was freed from large aggregates and centrifuged for 7 min at 650 × g, and the cell pellet was resuspended in DMEM (Sigma) supplemented with 10% FCS, 2% chick embryo extract, 1 mM glutamine (Sigma), 1000 U/ml penicillin (Sigma), and 50 μg/ml streptomycin (Sigma) (complete medium). The cells were counted in a hemocytometer and the cell density adjusted to 2 × 106 cells/ml. The cell suspension was plated in culture dishes or cover slips and incubated with a humidified 95% air/5% CO2 atmosphere at 37C. After 24 hr of incubation, the cells were washed three times with DMEM and maintained for 4 additional days with a change of the medium at 3 days of incubation.
L. amazonensis adhesion to or internalization by microglial cells or macrophages after 2-hr interactionsa
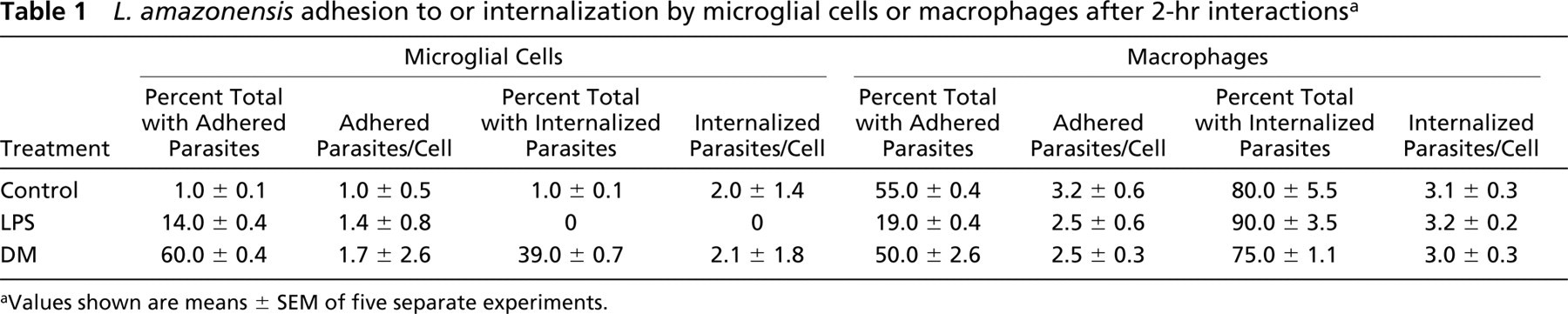
Values shown are means ± SEM of five separate experiments.
Macrophages
Murine macrophages were obtained from adult Swiss mice by harvesting of the peritoneal cavity, followed by plating of the cell suspension in 24-well culture dishes and incubation in DMEM in a humidified 95% air/5% CO2 atmosphere at 37C for 1 hr. Afterward, nonadherent cells were removed by washing and the adherent cells were cultured in complete medium. Macrophages were preincubated with 0.1 μg/ml DM or 1 μg/ml LPS at 37C for 24 hr and used for comparisons of adhesion to and/or internalization of L. amazonensis by microglial cells in mixed glial cultures after 2 hr of interaction of either mammalian cell with parasites (Table 1).
Parasite
L. amazonensis (MCAN/BR/94 DCB-16) were isolated from dogs with cutaneous lesions as amastigote forms and afterward maintained in Schneider medium (Sigma) supplemented with 10% FCS for 6–7 d. Within this period, the culture was followed at daily intervals to verify when the parasites transformed into promastigote forms and reached the stationary growth phase. The promastigote forms (Figure 1B) were used to infect the cultured cells at this phase.
Cell Identification
For identification of the microglia, the cultures were treated with either the BSI-B4 isolectin of Griffonia simplicifolia (Sigma) or with the F4–80 monoclonal antibody (Caltag Laboratories; San Francisco, CA). In the first case, the cells were incubated with 25 μg/ml BSI-B4 coupled to FITC at 37C for 1 hr or by binding of biotinylated BSI-B4 isolectin followed by reaction with horseradish peroxidase (HRP)-avidin (Sigma; EXTRA-2) and the peroxidase activity was revealed with H2O2 using 3-amino-9-ethyl carbazol (AEC; Sigma) as coupler (Figure 1A). For double staining of microglial cells, the cultures were also treated with the F4–80 primary antibody. Briefly, the samples were washed three times in PBS and incubated in blocking solution (10% normal rabbit serum diluted in PBS plus 1% BSA) plus 0.1% saponin for 1 hr at 37C, incubated with F4–80 diluted 1:10 at 37C for 1 hr, washed three times in PBS, and incubated with a Cy3-tagged rabbit anti-rat IgG secondary antibody diluted 1:300. Controls were treated by omission of the primary antibody. The nuclei of the cells were stained with 4',6-diamino-phenylin-dole (DAPI; 0.1 μg/ml) (Sigma). The samples on cover slips were mounted on slides with diazabicyclo[2.2.2] octane (DABCO) (Sigma) and analyzed with a Zeiss epifluorescence photomicroscope (Zeiss do Brasil, Sao Paulo, Brazil).
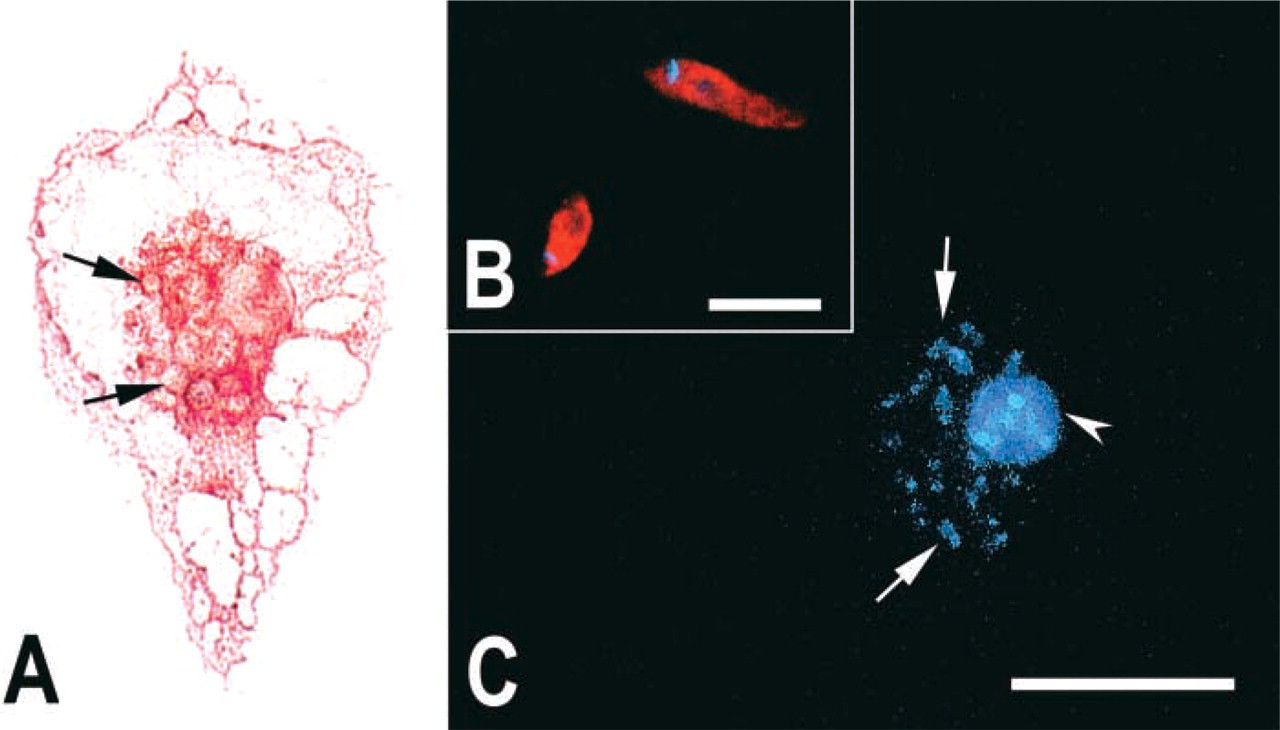
Phenotypic identification of infected microglia and Leishmania by glycoconjugates. (
For identification of the parasites, the cultures infected as described above or the promastigotes attached onto poly-
Surface Morphology of Infected Microglial Cells
Five-day-old cultures were pretreated for 24 h with 0.1 μg/ml DM or complete medium, washed three times with PBS, and then incubated with promastigote forms at 4C for 1 hr (parasite/microglia ratio = 10:1). DM-treated and untreated infected cells were rinsed, fixed for 1 hr in 2.5% glutaralde-hyde (Sigma), postfixed with 1% osmium tetroxide (Sigma) for 15 min, and dehydrated in acetone. Afterward, they were dried in CO2 in a critical point dryer (Balzers CPD030; Balzers Union, Lichtenstein) and coated with gold (Balzers MED 010). Samples were examined in a scanning electron microscope (Zeiss DSM 940).
Kinetic studies of Microglia–Leishmania Interaction
Mixed glial cultures were preincubated in DMEM/FCS with 0.1 μg/ml DM or 1 μg/ml LPS or maintained without treatment (control) at 37C for 24 hr. After that, the cultures were washed several times with serum-free DMEM and the total number of cells was estimated by counting several fields. Promastigote forms of L. amazonensis, previously centrifuged and suspended in serum-free DMEM, were added to the cultures to achieve a ratio of ≃10:1 parasites/glial cells, and the cell/parasite contact was maintained for 2 hr. After this period, cultures were rinsed with PBS to remove extracellular parasites, DMEM with 2% BSA was added, and the infection was followed at 37C for up to 72 hr, with fixation of infected cells at 0, 2, 4, and 10 hr after PBS rinsing (2, 4, 6, and 12 hr of interaction). Infected cells were fixed with Bouin's solution and stained with Giemsa. The percentage of microglial cells containing parasites attached or internalized was then determined by examining, in bright-field optics, randomly selected cells in at least 300 fields at 1000× magnification with a Zeiss photomicroscope (see Figures 3 and 4). To avoid missing internalized parasites that might have undergone shape or size changes, every microglial cell was analyzed in a through-focus mode.
DNA Degradation of Internalized Parasites
To approach the question of microglial killing of internalized parasites, a triple-labeling procedure was used. First, microglial cells in fixed cultures were identified by binding of biotinylated BSI-B4 isolectin followed by HRP-avidin (Sigma), and the peroxidase activity was revealed with H2O2 and AEC. After that, components of the parasites were detected with the 45D3 monoclonal antibody as described above. Finally, the infected cells were incubated with the TUNEL kit as described by the manufacturer (Boehringer, Mannheim, Germany).
For the TUNEL labeling, the samples on cover slips, previously fixed in 4% paraformaldehyde at room temperature, were permeabilized with 0.1% saponin (Sigma) on ice for 30 min. After washing, the samples were incubated with the TUNEL reaction mixture containing Terminal deoxyribonucleotide Transferase (enzyme solution) and fluorescein-dUTP (labeling solution) at 37C for 1 hr. The positive controls were treated by incubating the infected cells in the presence of DNase I (Sigma) and applying the TUNEL procedure. The cover slips were mounted on slides with DABCO and analyzed under bright-field and epifluorescence conditions.
Results
In phase-contrast optics or preparations stained by the Giemsa method, most cells (≃76%) in our 5-day cultures presented morphological features consistent with those of protoplasmic astrocytes (Moura Neto et al. 1985), occurring either as single cells or as small clusters of confluent cells (data not shown). Cells of varied morphologies (≃23%) were also found either on top of the astrocyte clusters or adherent to the plastic and were tentatively identified as microglia. This morphological identification was confirmed by phenotypic cell markers both before (data not shown) and after (Figure 1A) infection. Approximately 1% of the cells remained unidentified.
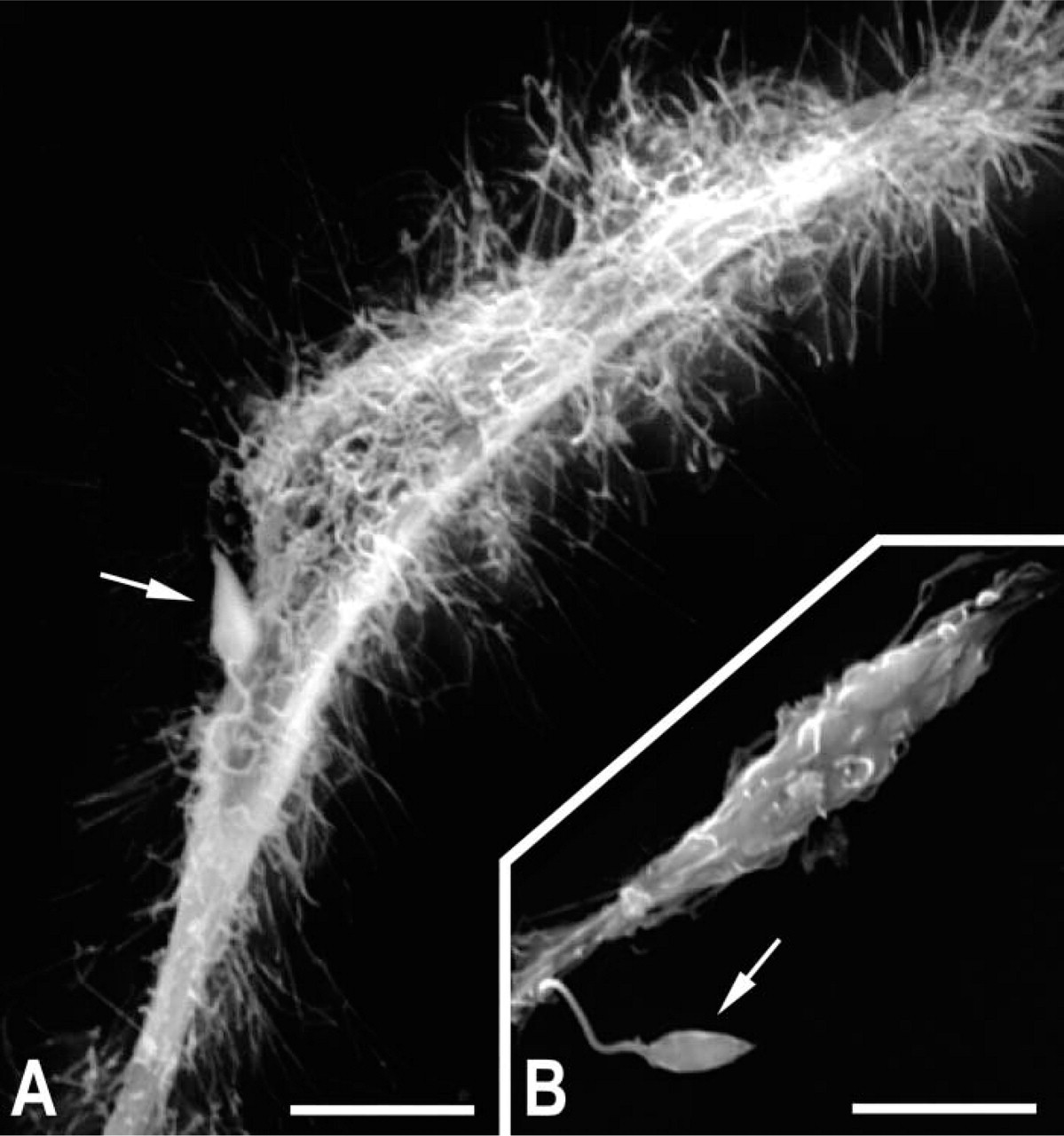
Ultrastructural analyses showing adhesion of promastigotes of L. amazonensis to DM-treated microglia (
Surface Morphology of Infected Microglial Cells
To demonstrate the in vitro influence of immune function mediators on the morphology of microglial cells, we used the macrophage function modulators DM and LPS during the interaction of microglia with promastigote forms of L. amazonensis. After pretreatment of the host cell for 24 hr, followed by washing and interaction with the parasite at 4C for 1 hr, scanning electron microscopy analysis showed numerous filopodia (Figure 2A) in DM-treated cells, whereas these projections were rarely observed in LPS-treated (data not shown) or control (Figure 2B) cultures, even in the presence of adhered parasites.
Quantitative Analysis of L. amazonensis Adhesion to and Internalization by Microglial Cells or Peritoneal Macrophages
Rare microglial cells (1% only) displayed parasites adhered to their surface, whereas adhesion to peritoneal macrophages occurred in more than half of these cells (Table 1, control). LPS or DM treatment of cultures showed a differential response for the two cell types. The treatment of microglial cells with LPS was effective in promoting the adhesion of parasites to their cellular surface, with such adhesion increasing from 1% in controls to 14% in the LPS-treated cells. By contrast, after LPS treatment of peritoneal macrophages, the percentage of cells with adhered parasites decreased from 55% in the control to 19% (Table 1). There were no major changes in the number of adhered parasites per microglial cell or per macrophage after treatment with LPS.
After DM treatment, there was a significant increase in the percentage of microglial cells with adhered parasites, increasing from 1% in the untreated cultures to 60% in the DM-treated cultures (Table 1). On the other hand, this increase was not observed in macrophages after the same treatment with DM, so that the percentage of cells with adhered parasites remained nearly identical to half of the cells. There were no major changes in the number of adhered parasites per microglial cell (Figure 3) or per peritoneal macrophage (Table 1) vis-à-vis the treatment with DM.
With respect to the internalization of parasites, similar to what was observed in adhesion assays, a very low number of untreated microglial cells presented intracellular parasites compared with untreated peritoneal macrophages (1% vs 80%; Table 1). No intracellular parasites were observed in microglial cells after treatment with LPS, whereas a very large percentage (90%) of macrophages contained intracellular parasites. The number of intracellular parasites per LPS-treated infected macrophage was similar to that in control cells.
The preincubation of microglial cells with DM favored parasite–cell interactions. Thus, the percentage of cells with intracellular parasites increased from 1% in the control to 39% after DM treatment (Table 1). However, the number of intracellular parasites per cell tended to remain unaltered in this short-term experiment. In the case of macrophages, we observed that 80% of the untreated cells already showed an average of 3.1 parasites per cell, whereas the percentage observed after treatment with DM was 75% with 3.0 parasites per cell (Table 1).

Giemsa-stained microglial cells from DM-treated (
Kinetics of Microglia Infection by L amazonensis
In preparations stained by the Giemsa procedure, we evaluated the kinetics of endocytosis of the promastigote forms of L. amazonensis in microglial cells maintained only in medium or preincubated with LPS or DM for 24 hr. The intracellular destiny of the parasites was analyzed after interaction with microglia for 2 hr, after which the cultures were washed and maintained in serum-free DMEM (2% BSA added), and the infection was followed for periods of 2, 4, 6, and 12 hr. In the cultures preincubated with LPS, we observed no internalized parasites or their residues in the host cell at any of the time intervals used (data not shown).
A significant number of internalized parasites was seen in DM-treated cells at 2 hr after infection (Figure 4B) compared with the untreated cells (Figure 4A). At 4 hr, stained profiles that could not be identified as viable parasites were observed in untreated cultures. In these untreated cells, there were structures in vacuoles distributed in different regions of the cytoplasm, but such structures were not clearly identified as parasites (Figure 4C). At the same time interval, in DM-treated cultures, several parasitophorous vacuoles with a large amount of cellular remains similar to killed parasites were observed in the microglial cytoplasm (Figure 4D), indicating a delay in the elimination of parasites in DM-treated cultures compared with untreated cultures. At 12 hr after infection, only vestiges of presumptive parasites were found in any of the cultures (Figures 4E and 4F).
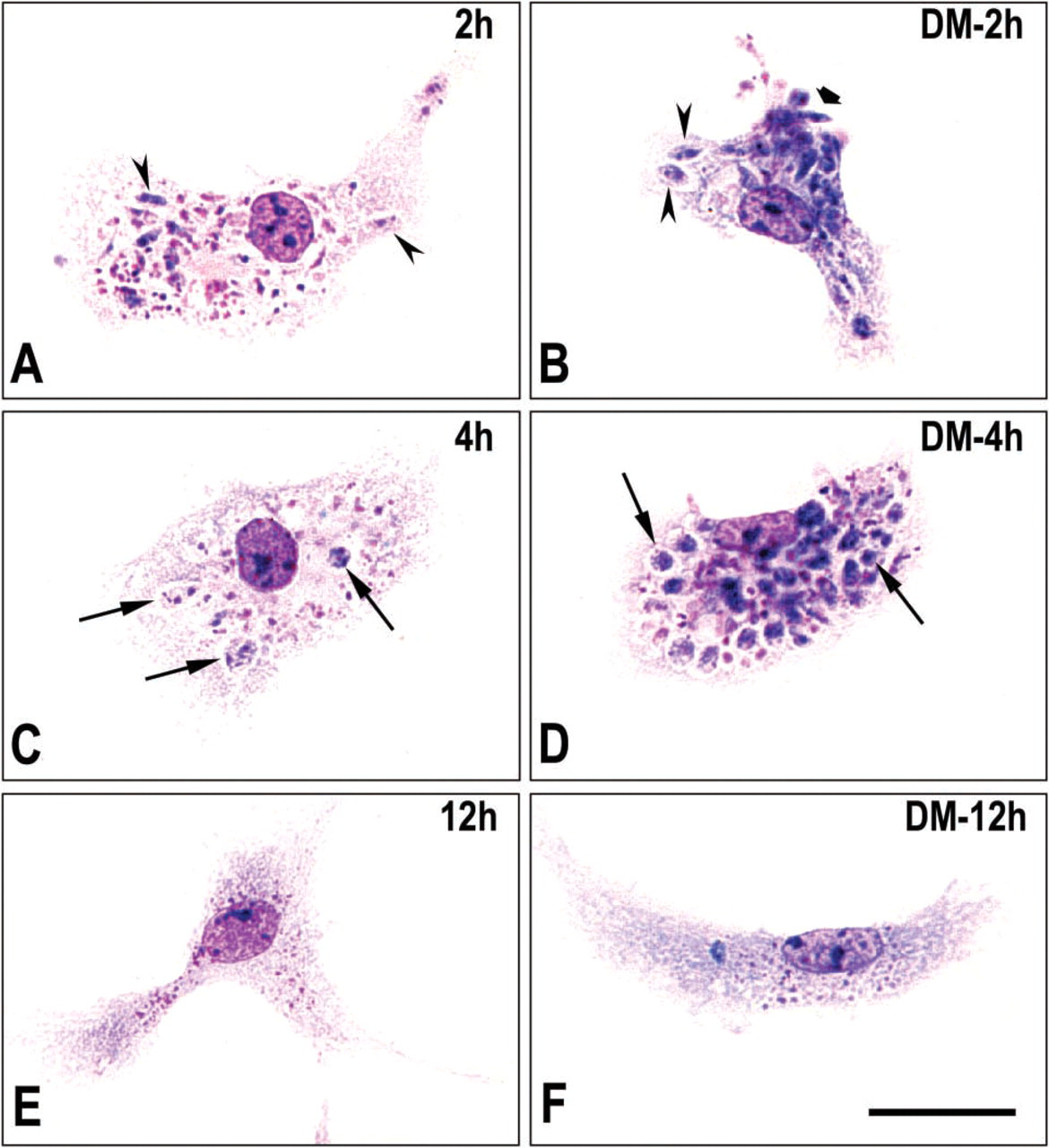
Kinetics of endocytosis of L. amazonensis by DM-treated microglia. Untreated control (
DNA Degradation of Internalized Parasites
To validate the findings of our study of the kinetics of L. amazonensis endocytosis by microglia, we used the TUNEL technique to evaluate the intracellular survival of the parasites. That was done through the identification of fragments of nuclear and kinetoplast DNA generated during the degradation and/or death process by digestion of the parasite by cells identified as microglia via BSI-B4 isolectin staining. We also used the (anti-LPG) 45D3 monoclonal antibody, which recognizes a characteristic LPG on the surface of Leishmania promastigotes (Lang et al. 1991) that is absent on amastigotes (Pimenta et al. 1991), for the localization of intracellular parasites or their residues during the infection of microglia. The promastigote forms of L. amazonensis, used for infection of the cultures, showed binding of the anti-LPG antibody throughout their surfaces (compare Figure 1B).
The LPG antigen and fragmented DNA of the parasite were detected at the 2-hr interval within both control cells and DM-treated microglia (data not shown). TUNEL + nuclei and kinetoplasts of the presumptive promastigotes colocalized with LPG+ membrane structures of the parasite. There was a larger number of these LPG-labeled profiles together with nuclei of dead parasites in cells treated with DM compared with control cells (data not shown). At 4 hr, LPG+ profiles and nuclei of the tagged parasites were numerous, with apparent aggregation of LPG+ profiles in the control cells (Figures 5A and 5B) and their punctual distribution close to the nucleus of the DM-treated cells (Figures 5C and 5D). At 6 hr, the amounts of parasite membrane profiles and fragmented DNA were noticeably reduced in relation to the previous times, but the distribution of both membrane remains and DNA fragments was not altered largely in either group (Figures 5E–5H). Similarly, there was a common tendency for the aggregation of the LPG+ structures in both control and DM-treated cells (Figures 5E–5H). At 12 hr after infection, no LPG+ profiles or TUNEL-labeled particles were detected in control microglial cells (Figures 5I and 5J), whereas rare LPG+ profiles (Figure 5K) but no TUNEL label (Figure 5L) were found in DM-treated cells.
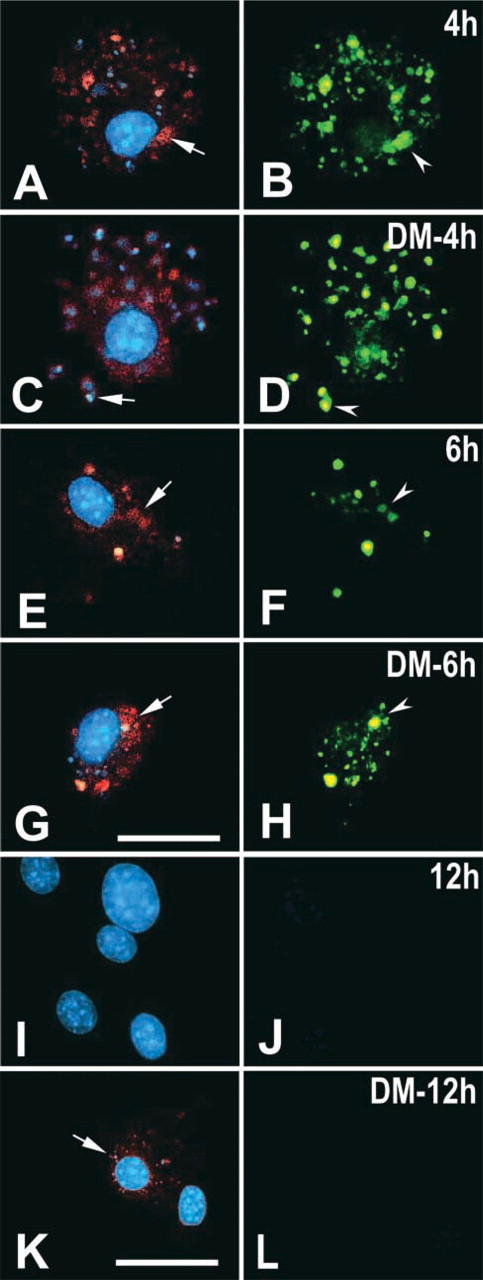
Kinetics of endocytosis of L. amazonensis by microglia in control cultures (
Discussion
Before proceeding to a discussion of our results, a few methodological comments are in order. In our experiments, we first tried to evaluate the infective potential of promastigote forms of L. amazonensis in astrocytes and/or microglial cells and verified that the protozoa adhered to and were internalized by microglia only. The use of primary glial mixed cultures was considered advantageous because microglial cells under this condition would be in an activation state different from that in pure cultures and, presumably, more akin to the in vivo situation (Glenn et al. 1989; Lee et al. 1992; Rezaie et al. 2002). It is known that, at least in some species, substances such as cytokines are secreted in mixed glial cultures and are able to cause morphological and phenotypical changes on microglia (Lauro et al. 1995; Liu et al. 1996).
Both LPS and DM treatments resulted in larger percentages of microglial cells with adhered parasites than for controls, possibly attributable to different mechanisms. Because LPS downregulates the mannose fucose receptor (MFR) (Marzolo et al. 1999), which is required for the establishment of intracellular parasitism in mononuclear phagocytes (Wilson and Pearson 1986), enhanced parasite adhesion in LPS-treated microglia may depend on the upregulation of molecules that do not trigger the internalization of parasites (Table 1).
In spite of the verification of a larger number of adhered parasites and increased percentage of cells that internalized parasites among those previously treated by DM compared with control cells, it was observed that apparently viable or intact parasites were no longer stained by the Giemsa method as soon as after 4 hr of interaction. This observation was reinforced by the TUNEL procedure, showing that in both untreated and DM-treated cultures there was fragmentation of the parasite nucleus and kinetoplast DNA as early as after 2–4 hr of interaction. The larger percentage of cells that internalized parasites in DM-treated cultures may derive from either or both of the following effects. First, the MFR could be upregulated by DM treatment, and this upregulation could lead to increased adhesion of parasites to the microglia surface and consequent internalization. This effect would be in agreement with the DM-induced MFR upregulation demonstrated in purified microglial cultures (Marzolo et al. 1999). Furthermore, preliminary studies of ours have shown that the addition of
A second effect would be DM treatment causing the downmodulation of inducible nitric oxide synthase and, thus, the reduction of nitric oxide synthesis and release. Again, it is known that cortisol (and possibly DM) inhibits the activation of microglia and decreases the production and release of nitric oxide and tumor necrosis factor-α (Drew and Chavis 2000), important cytotoxic and cytotoxicity-inducer molecules, respectively, for Leishmania (Roach et al. 1991). At present, we have no evidence that the apparently longer survival of L. amazonensis after DM treatment involves either the upregulation of the MFR receptor or the reduction of nitric oxide production by microglia in mixed cultures. The major astrocyte population of these cultures may express the MFR (Burudi et al. 1999) and the endothelial type of nitric oxide synthase (Wiencken and Casagrande 1999), making the identification of the source and/or the quantitative assessment of either MFR or nitric oxide ambiguous or unreliable. Alternative culture models are being tested in our laboratories to approach the questions of the mechanisms involved in phagocytosis and disposal of the parasite by microglia.
It cannot be ruled out that, as in monocytes (Ma et al. 2004), DM acts through the suppression of interleukin-12 production by microglia. In other words, DM would further decrease any remaining interleukin-12 production by microglia, which is already depressed by astrocytes (Aloisi et al. 1997), in mixed glial cultures. Clearly, additional work is necessary to investigate this issue.
The results obtained with the TUNEL procedure are compatible with the notion that DM treatment delays the killing and eventual disposal of Leishmania by microglia. More importantly, they emphasize that the microglial response to the parasite differs from that of peritoneal macrophages. Thus, there is an almost total absence of parasite residues in both DM-treated and untreated microglia after exposure of the cultures to promastigote forms for 12 hr.
In summary, microglial cells are highly effective in the elimination of Leishmania, at least in the particular case of mixed glial cultures. This cytotoxicity is apparently slowed by DM, indicating that microglia and other mononuclear phagocytes present both common and atypical functional features vis-à-vis intracellular parasites that must gain entrance into host cells to successfully complete their cell cycles. The mechanisms responsible for the microglial cytotoxicity of Leishmania remain to be determined.
Footnotes
Acknowledgements
Financial support for this work was provided by the Instituto Oswaldo Cruz, the Ministry of Science and Technology, the Brazilian Council for Science and Technology, and the Rio de Janeiro State Foundation for Research Support.
We thank Dr Carlos Alves for helpful suggestions on the detection of the LPG antigen. The excellent technical assistance of Bruno Avila and Sergio L. Carvalho is gratefully acknowledged. We also thank two unknown referees for their criticisms of and suggestions on a first version of this article.