
Abstract
Angiogenesis is a critical process in healing of myocardial infarcts, leading to the formation of highly vascular granulation tissue. However, effective cardiac repair depends on mechanisms that inhibit the angiogenic process after a mature scar is formed, preventing inappropriate expansion of the fibrotic process. Using a canine model of reperfused myocardial infarction, we demonstrated that maturation of the infarct leads to the formation of neovessels, with a thick muscular coat, that demonstrate distinct morphological characteristics. Many of these “neoarterioles” lack a defined internal elastic lamina and demonstrate irregular deposits of extracellular matrix in the media. Vascular mural cells in healing infarcts undergo phenotypic changes, showing minimal expression of desmin during the proliferative phase (1 hr occlusion/7 days reperfusion) but in the mature scar (8 weeks reperfusion) acquire a phenotype similar to that of vascular smooth muscle cells in control areas. Non-muscle myosin heavy chains A and B are induced in infarct endothelial cells and myofibroblasts, respectively, but are not expressed in neovascular mural cells. Recruitment of a muscular coat and formation of neoarterioles in mature scars may inhibit endothelial cell proliferation and vascular sprouting, stabilizing the infarct vasculature.
Keywords
T
VSMCs can express a range of phenotypes and respond to environmental cues by altering their expression of contractile proteins and by modifying their ability for protein synthesis. Vascular injury is associated with phenotypic modulation of smooth muscle cells, leading to increased expression of non-muscle myosin heavy chain (NMMHC) isoforms, such as NMMHC B/Smemb, in the vascular wall. Although structural and phenotypic alterations of VSMCs have been described in a variety of vascular lesions, the phenotype of vascular myocytes in healing wounds has not been systematically studied. Healing myocardial infarcts contain a large population of mesenchymal cells expressing α-smooth muscle actin (α-SMA) (Willems et al. 1994). The majority of these cells are phenotypically modulated myofibroblasts and do not express smooth muscle myosin (Frangogiannis et al. 2000b). As the wound matures, many infarct neovessels become coated with vascular mural cells, leading to the formation of a scar containing predominantly coated vessels (Ren et al. 2002). In this study we examined the progressive phenotypic changes of pericyte-coated vessels in infarcts and report significant changes in VSMC phenotype in mature scars. We describe for the first time dynamic changes in the morphological features of coated vessels that lead to a progressive increase in vessel wall thickness and the formation of a large number of vascular structures that lack an internal elastic lamina. Infarct maturation is associated with increased expression of desmin in the neovascular smooth muscle cells and with formation of vessels with large irregular deposits of matrix proteins in the forming media. In addition, we report for the first time induction of the non-muscle myosin heavy chain (NMMHC) A and B genes in the reperfused infarcted canine heart.
Materials and Methods
Ischemia/Reperfusion Protocols
All animal research protocols were approved by the Baylor College of Medicine Animal Research Committee. Healthy mongrel dogs (15–25 kg) of either sex were surgically instrumented as previously described (Frangogiannis et al. 1998a,b; Frangogiannis et al. 2000a). Anesthesia was induced IV with 10 mg/kg methoexital sodium (Brevital; Eli Lilly, Indianapolis IN) and maintained with the inhalational anesthetic isoflurane (Anaquest; Madison, WI). A midline thoracotomy provided access to the heart and mediastinum. A hydraulically activated occluding device and a Doppler flow probe were secured around the circumflex coronary artery just proximal or just distal to the first branch. Indwelling catheters placed in the right atrium, left atrium, and femoral artery allowed blood sampling and pressure monitoring as needed. After surgery the animals were allowed to recover for 72 hr before coronary occlusion. Ischemia/reperfusion protocols were performed as previously described (Frangogiannis et al. 1998a). Coronary artery occlusion was achieved by inflating the coronary cuff occluder until mean flow in the coronary vessel was zero as determined by the Doppler flow probe. At the end of 1 hr, the cuff was deflated and the myocardium was reperfused. Reperfusion intervals ranged from 5 hr to 8 weeks. Circumflex blood flow, arterial blood pressure, heart rate, and ECG (standard limb II) were recorded continuously. Analgesia was accomplished with IV pentazocine (Talwin; Winthrop Pharmaceuticals, New York, NY) 0.1–0.2 mg/kg. After the reperfusion periods, hearts were stopped by rapid IV infusion of 30 mEq of KCl and removed from the chest for sectioning from apex to base into four transverse rings, 1 cm in thickness. The posterior papillary muscle and the posterior free wall were identified. Tissue samples were isolated from infarcted or normally perfused myocardium on the basis of visual inspection. Myocardial segments were fixed for histological analysis in B∗5 without formalin (Beckstead 1994) and embedded in paraffin. Duplicate adjacent samples were processed for blood flow determinations using radiolabeled microspheres. The presence of a myocardial infarct was based on light microscopic examination of hematoxylin-eosin-stained tissue sections by findings of replacement of cardiomyocytes with granulation tissue or scar. Samples of control tissues were taken from the anterior septum and had normal blood flow during coronary occlusion.
Animals included in the study underwent 1 week (
In addition, four healthy mongrel dogs were sacrificed and the segments from the left ventricle and the following vessels were obtained: thoracic ascending aorta, thoracic descending aorta, carotid, subclavian, femoral, and coronary arteries, superior vena cava, jugular vein, and femoral vein. The samples were fixed in B∗5 without formalin and embedded in paraffin.
Tissue Processing, Histology, and Immunohistochemistry
Histological samples were fixed in B∗5 fixative and embedded in paraffin. Sequential 3–5-μ sections were cut by microtomy. Collagen and elastin fibers were identified by Verhoeff-van Gieson (VVG) staining. Immunostaining was performed using the ELITE rabbit, goat, or mouse kit (Vector Laboratories; Burlingame CA). Briefly, sections were pretreated with a solution of 3% H2O2 to inhibit endogenous peroxidase activity and incubated with 2% horse serum to block nonspecific protein binding. Then they were incubated with the primary antibody for 2 hr at room temperature. After rinsing with PBS, the slides were incubated for 30 min with the secondary antibody. The slides were rinsed with PBS and incubated for 30 min in ABC reagent (Hsu et al. 1981). Peroxidase activity was detected with diaminobenzidine (DAB; Vector). Slides were counterstained with eosin. The following primary antibodies were used for IHC: mouse anti-α-SMA (Frangogiannis et al. 2000b), mouse anti-desmin (both from Sigma; St Louis MO), mouse anti-smoothelin (Abcam; Cambridge, MA), mouse anti-CD31 (Dako; Carpinteria, CA) (Frangogiannis et al. 2000b), mouse anti-collagen type III (ICN; Aurora, OH) (Frangogiannis et al. 2003), and rabbit anti-NMMHC A and B (Covance; Berkeley CA).
Quantitative Morphometry and Statistical Analysis
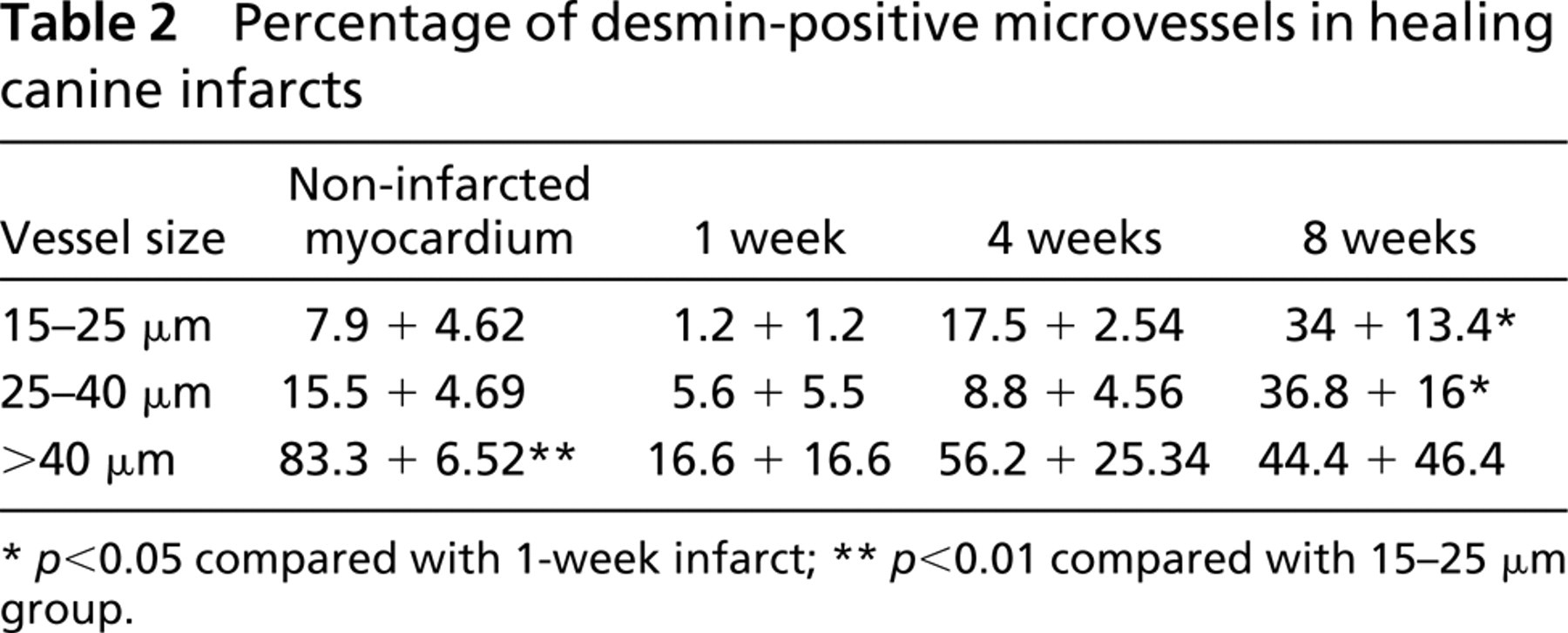
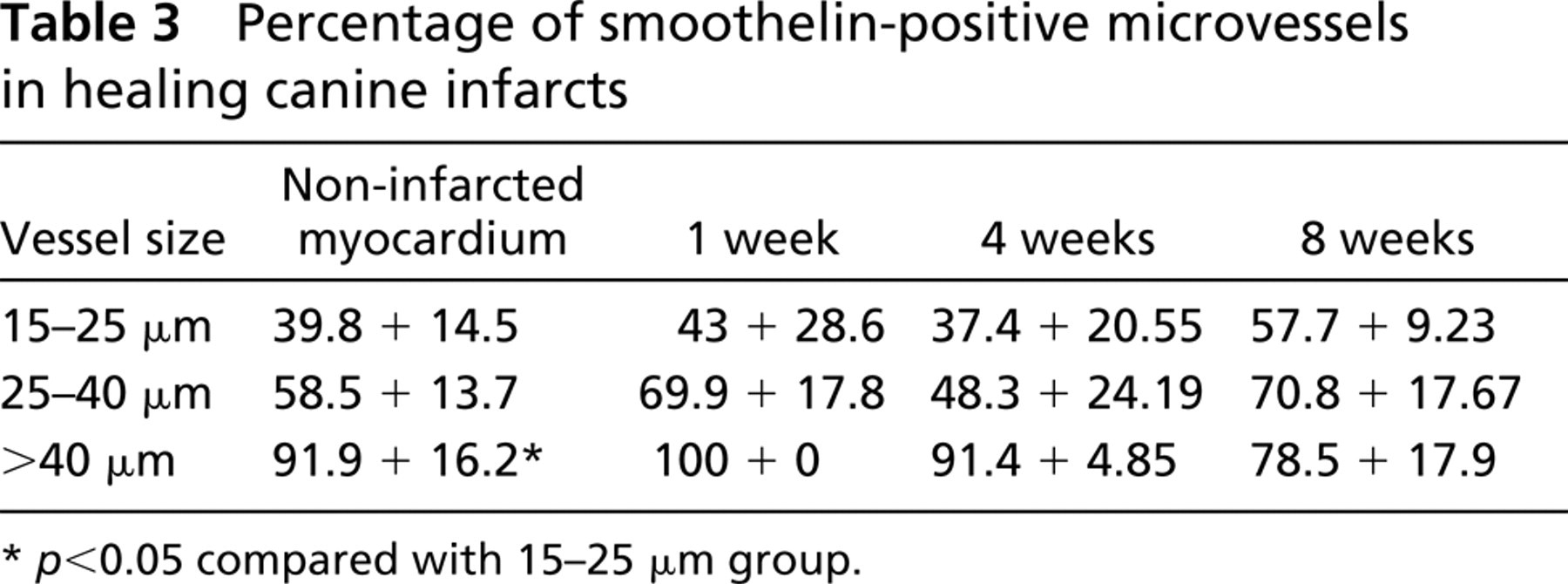
At least two ischemic and two control samples were studied from each experiment. Stained slides were examined with a Zeiss Axioskop microscope and photographed with a Zeiss digital camera. For quantitation of the arteriolar density, all coated vessels with a diameter >15 μm were counted in infarcted and control areas and the density was expressed as vessels/mm2. In addition, the density of small (diameter 15–40 μm) and large arterioles (diameter 40 μm) was quantitated. Eight different fields from three control and ischemic segments from each experimental group (1 week, 4 weeks, and 8 weeks of reperfusion) were analyzed. Mean arteriolar wall thickness was quantitated for each field based on all arterioles seen in cross-section (major:minor axis <1.4). Staining for desmin, an intermediate filament protein, and smoothelin, a cytoskeletal protein expressed by differentiated smooth muscle cells, was used to assess the phenotype of vascular mural cells in healing infarcts. The percentage of desmin-and smoothelin-positive arterioles in infarcted and non-infarcted samples was quantitated by examining serial sections stained for α-SMA, desmin, and smoothelin. Segments from three different experiments (control and infarcted) from each group were used for analysis. All α-SMA-positive coated vessels in the fields examined were identified, classified into one of three groups based on diameter (15–25 μm, 25–40 μm, >40 μm) and examined for desmin and smoothelin staining. Negative staining was defined as complete absence of desmin or smoothelin immunoreactivity in the vascular wall. Statistical analysis was performed using ANOVA followed by
mRNA Extraction and Northern Hybridization
RNA isolation from myocardial tissue segments was performed using the acid guanidinium-phenol-chloroform procedure. RNA (20 μg) was electrophoresed in 1% agarose gels containing formaldehyde and then transferred to a nylon membrane (Gene Screen Plus; New England Nuclear. Boston, MA) by standard procedures. The membranes were hybridized in QuikHyb (Stratagene; La Jolla, CA) at 68C for 2 hr with 1 × 106 dpm random hexamer 32P-labeled cDNA probes for NMMHC-A and NMMHC-B (Simons et al. 1991) (a generous gift from Dr. Adelstein, National Institutes of Health). Filters were washed with 2 × SSPE at 68C for 20 min, with 1 × SSPE + 1% SDS at 68C for 15 min twice, and with 1 × SSPE at 21C for 15 min with constant shaking and exposed to Hyperfilm (Amersham; Arlington Heights, IL).
Results
Phenotypic Characteristics of Normal Canine Vessels
Canine vascular smooth muscle cells exhibited intense immunoreactivity for α-SMA. In elastic arteries such as the aorta, a large but variable proportion of smooth muscle cells stained for desmin and smoothelin (data not shown). In large muscular arteries, such as the femoral and coronary (Figures 1A–1C), most smooth muscle cells were desmin- and smoothelin-positive. Staining for both desmin and smoothelin was also intense in smaller muscular arteries. In the cardiac vasculature, intramyocardial coronary arteries (diameter >100 μm) invariably expressed desmin and smoothelin. In contrast, desmin and smoothelin immunoreactivity in myocardial arterioles was dependent on the size of the vessels, with large arterioles (>40 μm diameter) more often expressing desmin and smoothelin than smaller arterioles. Desmin staining in myocardial arterioles was often more prominent in the outer media (Figure 1E). Desmin and smoothelin staining was practically absent in pericyte-coated capillaries and in small veins and venules (Figures 1D–1F).
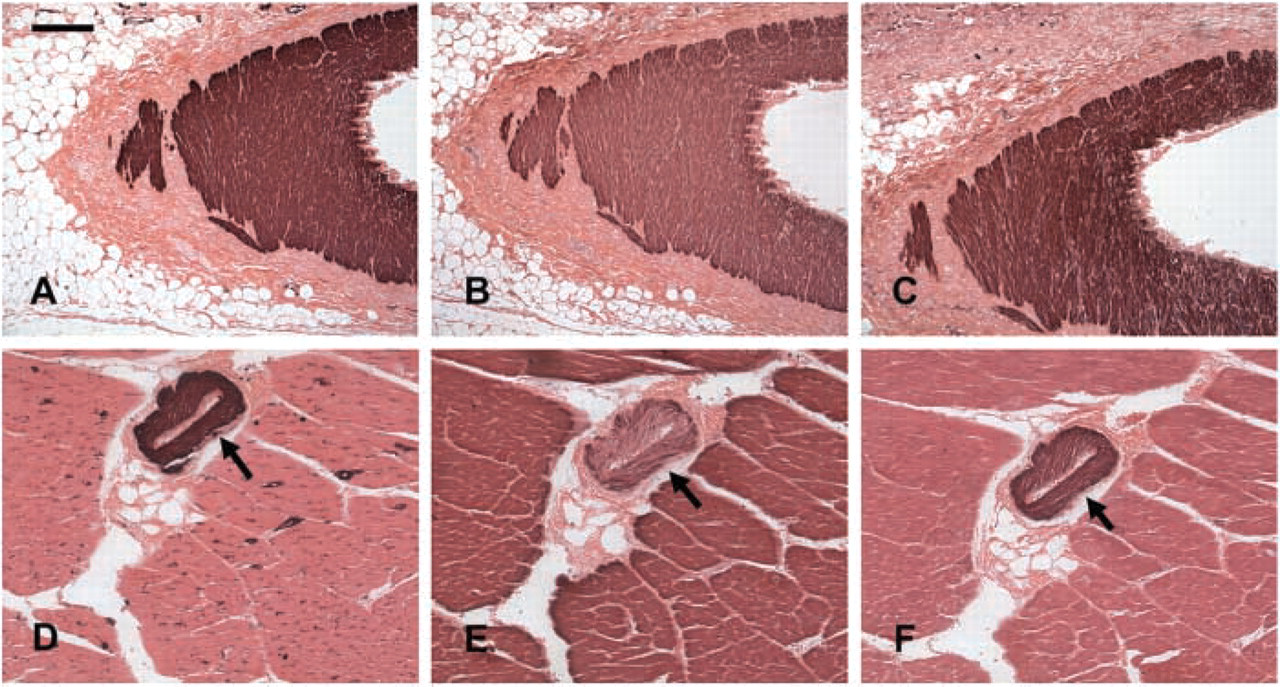
Expression of α-SMA, desmin, and smoothelin in canine epicardial coronary artery (
Morphological Characteristics of Arterioles in the Infarct Vasculature
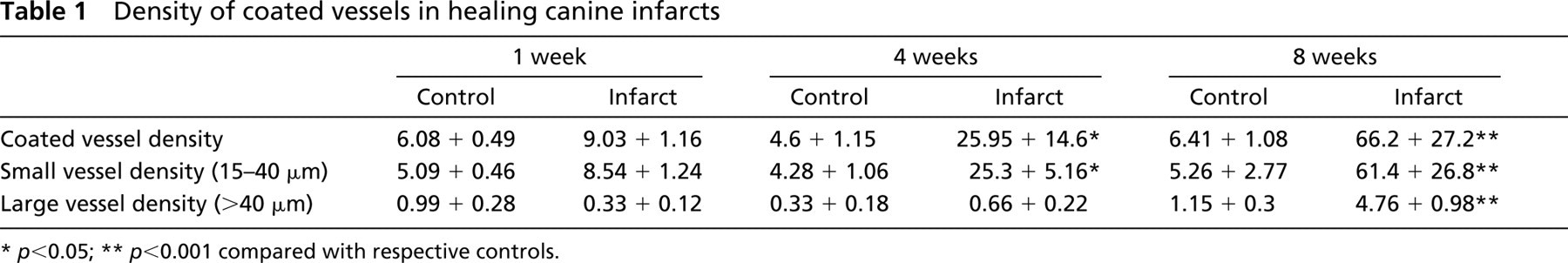
Infarcted canine hearts showed areas of cardiomyocyte replacement with granulation tissue after 7 days of reperfusion. α-SMA staining identified a large number of spindle-shaped myofibroblasts in the healing canine myocardium (Figure 2). In addition, α-SMA immunoreactivity was localized in vascular smooth muscle cells and pericytes coating microvessels of the infarcted heart. Maturation of the infarct was accompanied by a progressive increase in the number of vessels with a muscular coat (Table 1) (7 day reperfusion 9.03 + 3.27 vessels/mm2; 4 weeks 25.95 + 14.61 vessels/mm2; 8 weeks 66.21 + 27.26 vessels/mm2). The majority (>90%) of coated vessels in the healing infarcts had a diameter of less than 40 μm (Figure 3). However, in mature scars the mean wall thickness of coated vessels increased significantly (8 weeks 9.3 + 0.22 μm vs 1 week 6.13 + 1.14 μm;
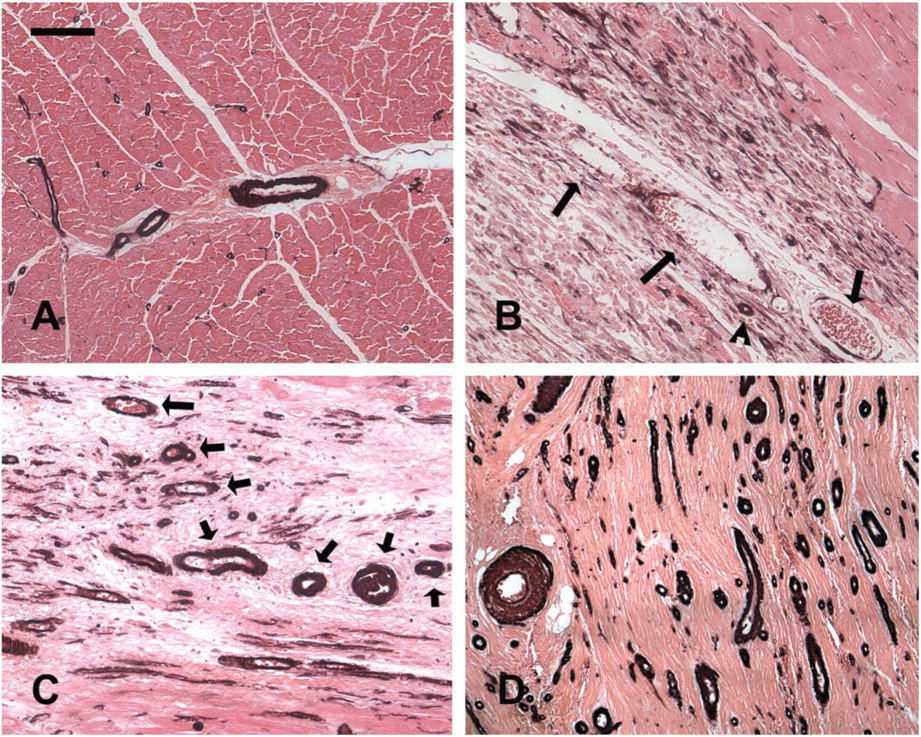
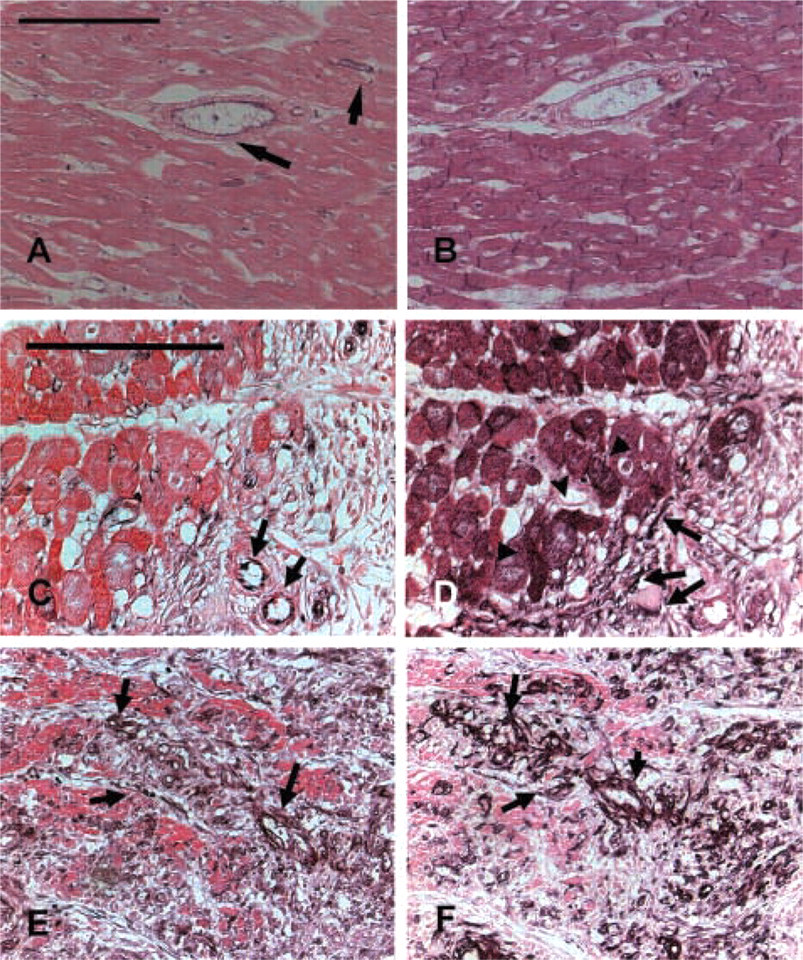
Acquisition of a muscular coat by neovessels in healing canine infarcts. α-SMA IHC in non-infarcted areas identifies arteriolar smooth muscle cells and pericytes (
The morphology of the infarct vasculature changed significantly over the course of healing. After 4 weeks of reperfusion, vessels with an irregular thin media were noted, appearing to actively recruit α-SMA positive mural cells (Figures 3A–3C). In the mature scar, the fully coated infarct vessels had distinct morphological features compared with control myocardial arterioles from the same experiment. Many had no internal elastic lamina (IEL) (Figures 3D–3I) or exhibited irregular depositions of collagen and elastin in the media (Figures 3J–3O). Some of the larger arterioles had a discontinuous tunica media showing a thin inner layer of mural cells around the vascular lumen separated from the thicker outer media by ECM (Figures 3J–3O).
Phenotypic Characteristics of Vascular Mural Cells in the Infarct
We hypothesized that the vascular smooth muscle cells coating the infarct neovessels may undergo a maturation process, leading to a progressive increase in expression of desmin and smoothelin in the vascular wall. In the non-infarcted myocardium, desmin expression in the arteriolar media was dependent on the size of the vessel, with larger arterioles (>40 μm) showing a significantly higher percentage of desmin-positive vessels than smaller arterioles (15–40 μm) (
Density of coated vessels in healing canine infarcts
Expression of NMMHC-A and NMMHC-B in Dog Infarcts
In the control canine heart, Northern hybridization with the NMMHC-A and -B cDNA clones resulted in the detection of two distinct bands, a 7.5-kb band corresponding to the NMMHC transcripts and a smaller band, representing cross-hybridization of the probes with cardiac muscle myosin heavy chain, as described previously in rat heart by Simons et al. (1991). Canine infarcts exhibited significant induction of NMMHC-A mRNA after 5 hr of reperfusion. NMMHC-A mRNA levels peaked after 5–7 days of reperfusion. At this stage, many segments exhibited loss of the cardiac muscle myosin band, suggesting extensive infarction and cardiomyocyte replacement (Figure 5). A modest upregulation of NMMHC-B mRNA expression was detected in infarcted myocardial segments after 5 days of reperfusion.
NMMHC protein localization was examined with IHC in infarcted and non-infarcted canine tissue. In control canine myocardium, NMMHC-A was predominantly expressed in the vascular endothelium (Figure 6A), whereas NMMHC-B was immunolocalized in the intercalated disks of cardiomyocytes (Figure 6B). Normal myocardial arterioles and small intramyocardial arteries showed little expression of the NMMHC isoforms. In the infarct, NMMHC-A immunoreactivity was predominantly localized in the vascular endothelium but not in myofibroblasts or vascular smooth muscle cells after 7–28 days of reperfusion (Figure 6C). NMMHC-B staining was noted in cardiomyocytes of the border zone and in spindle-shaped mesenchymal cells of the infarcted area. Border zone cardiomyocytes exhibited diffuse cytoplasmic immunoreactivity for NMMHC-B (Figure 6D), a pattern resembling the staining previously described in embryonic cardiac myocytes (Murakami et al. 1993; Takeda et al. 2000). Many mesenchymal cells expressing NMMHC-B were identified as α-SMA-positive myofibroblasts. In contrast, the mural cells of coated vessels had minimal expression of NMMHC isoforms (Figures 6C, 6E, and 6F).
Discussion
Vascular lesions, such as atherosclerosis and post-angioplasty restenosis, are often associated with extensive alterations in VSMC phenotype (Shanahan and Weissberg 1998; Hao et al. 2003). In both these pathological processes, VSMCs undergo a transition from a “contractile” to a “synthetic” phenotype and acquire proliferative potential (Campbell and Chamley-Campbell 1981; Nobuyoshi et al. 1991). In addition, both atherosclerotic and restenotic lesions are associated with alterations in smooth muscle myosin heavy chain isoform expression (Aikawa et al. 1993; (De Leon et al. 1997) and induction of the NMMHC isoforms.
Although a number of studies have examined the alterations in smooth muscle cell phenotype after vascular injury, little information is available on the fate of smooth muscle cells in healing wounds. We have previously identified mature smooth muscle cells (expressing both α-SMA and the smooth muscle myosin isoforms SM1 and SM2) and activated myofibroblasts (expressing α-SMA but not SM1 and SM2, markers of mature smooth muscle cells) in healing canine infarcts (Frangogiannis et al. 2000b). Mature smooth muscle cells were found in the media of infarct arterioles, whereas myofibroblasts are predominantly located in the infarct border zone. The present study describes the morphological and phenotypical changes of the infarct arterioles, vascular structures with a potential role in scar maturation and stabilization.
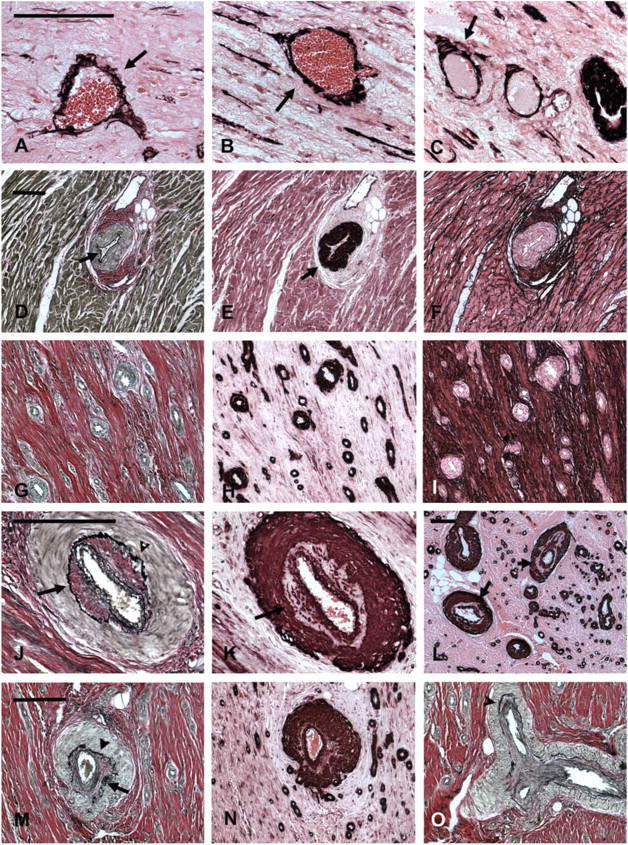
Morphological characteristics of “neoarterioles” in healing infarcts. (
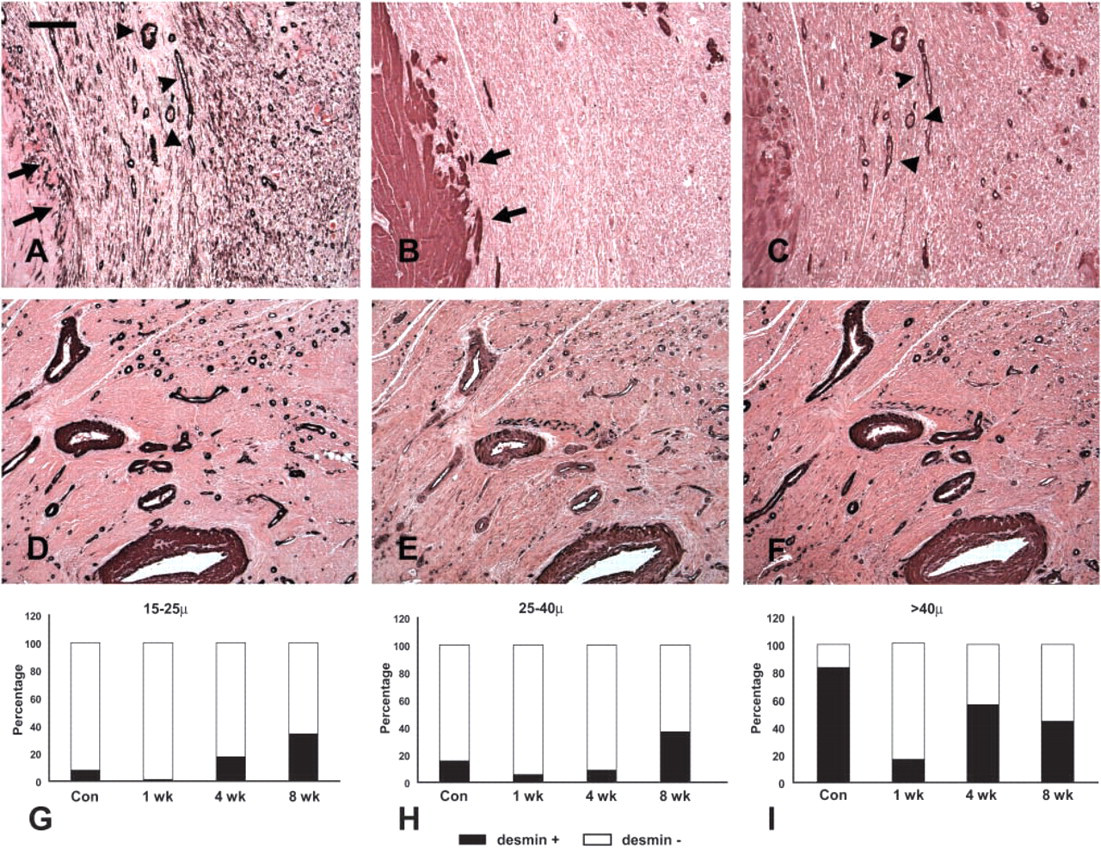
Expression of desmin and smoothelin by vascular mural cells in healing infarcts. (
Percentage of desmin-positive microvessels in healing canine infarcts
Morphological Characteristics of the Normal Canine Vasculature
To understand the structure and phenotype of infarct vessels, we first systematically examined the morphological characteristics of normal canine vessels. We used staining for the cytoskeletal proteins smoothelin, a marker of contractile smooth muscle cells (van der Loop et al. 1996), and desmin, to determine the phenotypic characteristics of VSMCs in normal dog vessels. Canine muscular arteries had higher expression of desmin and smoothelin than elastic arteries. In the cardiac vasculature, large arterioles were more likely to be desmin-positive than small arterioles (Figure 1). Smoothelin and desmin expression was absent in capillaries, venular pericytes, and small veins but was often observed in large venous SMCs. These findings are similar to the previously reported pattern of localization of desmin and smoothelin in human vessels (Johansson et al. 1997,1999; van der Loop et al. 1997).
The Infarct “Neoarteriole”: Morphology and Phenotype
During the proliferative phase of healing, wound angiogenesis is important to provide the highly cellular and metabolically active granulation tissue with oxygen and nutrients (Frangogiannis et al. 2002b). At this stage, enlarged pericyte-poor vessels (Pettersson et al. 2000; Ren et al. 2002), many capillaries, and abundant myofibroblasts are noted in the healing infarct; these cells are responsible for collagen deposition in the wound (Cleutjens et al. 1995). As the infarct matures, the highly vascular granulation tissue is replaced by a collagen-rich scar. The mature scar exhibits a relatively low capillary density but a high arteriolar density and a large number of pericyte-coated vessels (Ren et al. 2002). We coined the term “neoarteriole” to describe these newly formed coated infarct vessels, which have certain unique morphological characteristics (Figures 2 and 3). In contrast to normal myocardial arterioles, many neoarterioles do not have a defined IEL but often exhibit irregular deposits of matrix in the media (Figure 3), which is sometimes divided into two separate layers. The lack of an IEL in infarct neoarterioles may reflect continuous vascular remodeling. In addition, because of the role of the IEL as a barrier separating the intima and the media of the vascular wall, its absence may result in increased outward diffusion of plasma macromolecules into the arteriolar media (Penn et al. 1994). Wall thickness progressively increases as the scar matures and occasional bizarre vascular structures with multiple walls are noted. Vessels with an irregular media are also found, suggesting a continuous coating process during infarct maturation (Figure 3). Considering the low metabolic needs of the mature scar and the small number of capillaries, it is likely that coating of infarct neovessels with mural cells does not serve to create effective conduits of blood; it rather represents a mechanism necessary for inhibition of the angiogenic process. This view is supported by previous experiments (Connelly et al. 1989) demonstrating that myocardial blood flow in reperfused rabbit infarcts 3 weeks after infarction is significantly lower than blood flow during the early post-reperfusion period. Recruitment of mural cells by the infarct neovasculature may be important for stabilization of the wound by inhibiting endothelial cell proliferation and angiogenesis and by protecting the neovessels from regression (Benjamin et al. 1998).
Percentage of smoothelin-positive microvessels in healing canine infarcts
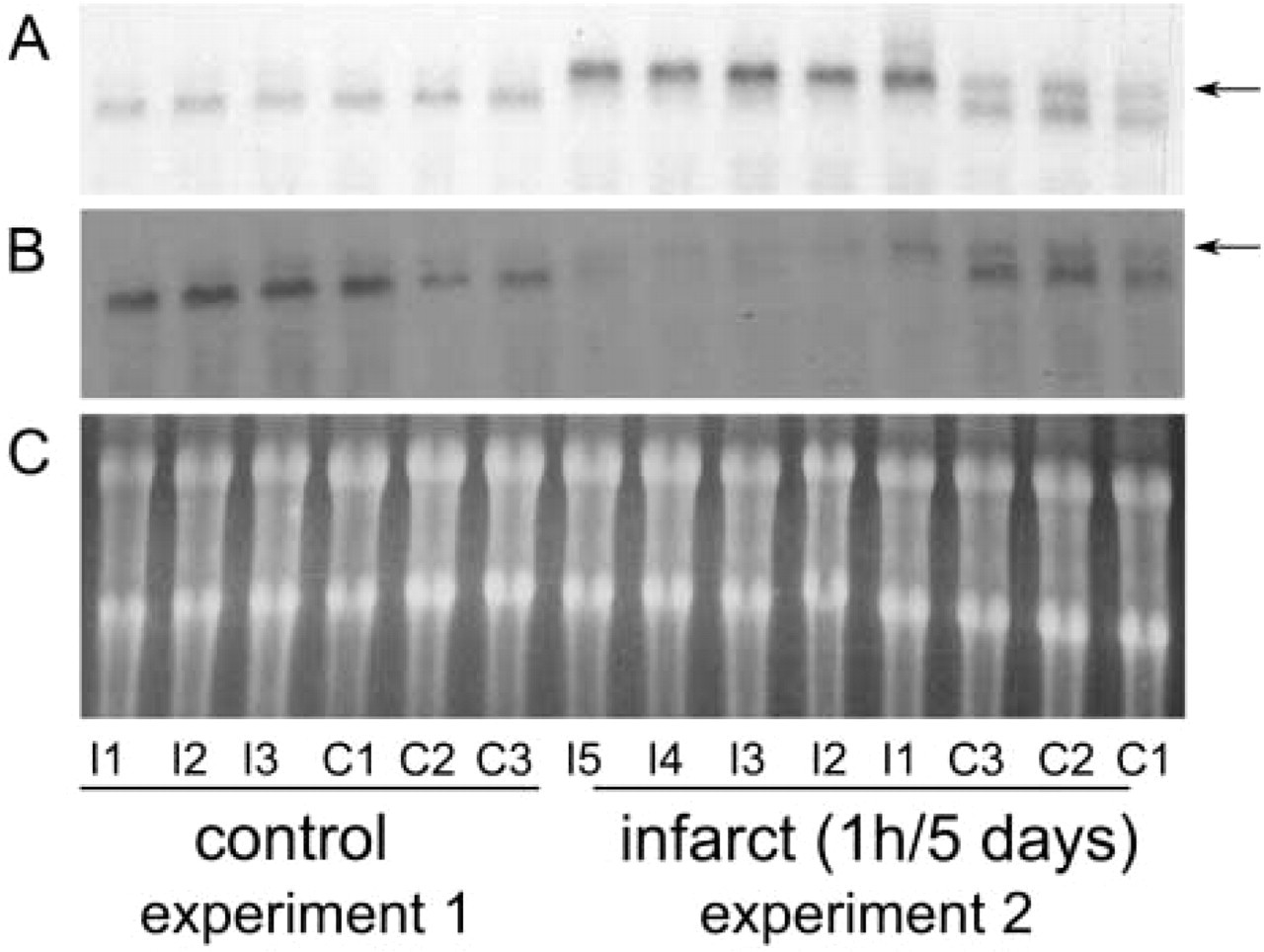
NA expression in canine infarcts. Northern hybridization of representative experiments of 1-hr circumflex coronary occlusion and 5 days of reperfusion. Segments were identified as I and C depending on their location (I, segments in the territory perfused by the circumflex coronary artery; C, segments from the anterior septum, not perfused by the circumflex coronary artery). Experiment 1 underwent instrumentation and inflation of the occluder but had no evidence of myocardial infarction (probably because of the presence of extensive collateral circulation), showing normal segmental blood flow during occlusion by the method of radiolabeled microspheres. Experiment 2 had extensive infarction; all segments labeled as I had blood flow <25% of control (
NMMHC immunoreactivity in canine hearts. (
VSMC Maturation in the Infarct
VSMCs demonstrate remarkable plasticity and undergo significant phenotypic changes, depending on the microenvironment. In atheroslerotic plaques, the cytoskeletal features of VSMCs are modified compared with resident medial smooth muscle cells (Kocher and Gabbiani 1986). These cells acquire a dedifferentiated phenotype as far as the cytoskeleton is concerned, showing high expression of vimentin and a relatively low expression of desmin (Gabbiani et al. 1984). Using IHC staining for desmin and smoothelin, we investigated the phenotypic changes of vascular mural cells in dog infarcts. During the proliferative phase of healing (7 days after infarction), the majority of coated vessels contained a desmin-negative media. In contrast, in mature scars (8 weeks after infarction), coated vessels had desmin content comparable to that of non-infarcted hearts (Figure 4; Table 2). However, in contrast to non-infarcted myocardium, in which arteriolar desmin expression was found predominantly in larger arterioles (>40 μm), mature infarcts had similar desmin expression regardless of size. Although the function of desmin in vascular smooth muscle cells is not completely understood, its absence results in lower distensibility (Lacolley et al. 2001) and microvascular dysfunction (Loufrani et al. 2002). The increase in the number of desmin-positive microvessels during infarct maturation may reflect differentiation of the vascular mural cells and may affect the viscoelastic properties of the infarct vasculature. Surprisingly, smoothelin, a cytoskeletal protein found in mature contractile VSMCs, was similarly expressed in vessels from infarcted and non-infarcted areas (Figure 4; Table 3). Although this may suggest that infarct mural cells express certain smooth muscle cell differentiation markers at an early stage, it may also reflect the lack of sensitivity of IHC methods in detecting different levels of protein expression.
Non-muscle Myosin Heavy Chain Isoforms in the Infarct
Non-muscle myosins of the myosin II superfamily are ubiquitously expressed motor proteins, potentially involved in a variety of physiological tasks, including cell migration and cytokinesis (Spudich 1989; Lofgren et al. 2003). Cultured smooth muscle cells, while actively growing, and embryonic smooth muscle tissues predominantly express NMMHC (Kawamoto and Adelstein 1987). In addition, atherosclerotic rabbit aortas express significant amounts of NMMHC isoforms, suggesting a dedifferentiation process towards an embryonic smooth muscle cell phenotype (Zanellato et al. 1990). We have previously demonstrated that healing canine myocardial infarcts (Frangogiannis et al. 2000b) and myocardial segments from patients with ischemic cardiomyopathy (Frangogiannis et al. 2002a; Frangogiannis 2003) contain a large number of NMMHC-B/Smemb-expressing mesenchymal cells. In healing infarcts these cells were predominantly located in the infarct border zone and were identified as myofibroblasts by their expression of α-SMA. Vascular mural cells were SM1- and SM2-positive but exhibited no staining for SMemb. In the present study we examined mRNA and protein expression of both NMMHC isoforms in healing canine infarcts. NMMHC-A expression was noted in the non-infarcted myocardium and was predominantly localized in the microvascular endothelium (Figure 6). Significant NMMHC-A mRNA upregulation was found in healing infarcts (Figure 5), and NMMHC-A protein was localized in the endothelium of infarct neovessels (Figure 6C). Myofibroblasts and vascular mural cells did not demonstrate significant NMMHC-A immunoreactivity. NMMHC-B protein was immunolocalized in the intercalated disks of cardiomyocytes in the non-infarcted heart. This is consistent with previously reported findings in human and murine tissues (Takeda et al. 2000). In infarcted myocardial segments, a modest upregulation of NMMHC-B mRNA was noted. IHC identified α-SMA-expressing myofibroblast-like cells as the main source of NMMHC-B protein expression in the infarct, confirming our previously reported findings with a different antibody to NM-MHC-B/SMemb (Frangogiannis et al. 2000b). Vascular mural cells coating infarct neovessels did not stain for NMMHC-B. Interestingly, many border zone cardiomyocytes exhibited heavy and diffuse cytoplasmic staining for NMMHC-B, in sharp contrast to the normal cardiomyocytes, which show localized NMMHC-B expression in the intercalated disks (Figure 6D). This pattern of staining has been previously described in embryonic hearts (Takeda et al. 2000). These cells may be resident cardiomyocytes that acquired an embryonic phenotype, or may represent a subset of stem cells expressing high levels of NMMHC-B.
Acquisition of a Muscular Coat: an Important Step for the Formation of a Mature Scar
Our study describes an important aspect of infarct healing, reporting the formation of neoarterioles in the wound. These vascular structures result from recruitment of vascular mural cells by infarct neovessels and exhibit distinct morphological features. During the proliferative phase of healing, highly vascular granulation tissue with a large number of capillaries is formed and may serve to supply the metabolically active infarct with nutrients and oxygen. However, as the scar matures, the metabolic needs of the infarct decrease and most capillaries regress as the remaining microvessels become coated with vascular mural cells. Formation of neoarterioles in the healing infarct may represent an important regulatory step in stabilizing the infarct by suppressing the angiogenic process. This investigation raises interesting questions regarding the origin of the vascular mural cells and the signals responsible for their recruitment. Blood-derived progenitors or resident myocardial mesenchymal cells may be the source of vascular myocytes coating infarct microvessels. PDGF- and endoglin-mediated pathways (Lindahl et al. 1997; Li et al. 1999) may be important in mediating recruitment of mural cells by the infarct endothelium. Mechanistic studies, using murine models of myocardial infarction, will be required to address these intriguing questions.
Footnotes
Acknowledgements
Spported by NIH Grant HL-42550, by a Grant-in-Aid from the American Heart Association Texas Affiliate, and by the DeBakey Heart Center.
We wish to thank Peggy Jackson, Alida Evans, and Stephanie Butcher for their outstanding technical assistance, and Sharon Malinowski and Connie Mata for editorial assistance with the manuscript.