
Abstract
Myocardial infarction (MI) is associated with an angiogenic response, critical for healing and cardiac repair. Using a canine model of myocardial ischemia and reperfusion, we examined the structural characteristics of the evolving microvasculature in healing MI. After 7 days of reperfusion, the infarcted territory was rich in capillaries and contained enlarged, pericyte-poor “mother vessels” and endothelial bridges. During scar maturation arteriolar density in the infarct increased, and a higher percentage of microvessels acquired a pericyte coat (60.4 ± 6.94% after 28 days of reperfusion vs 30.17 ± 3.65% after 7 days of reperfusion; p<0.05). The microvascular endothelium in the early stages of healing showed intense CD31/PECAM-1 and CD146/Mel-CAM immunoreactivity but weak staining with the Griffonia simplicifolia lectin I (GS-I). In contrast, after 28 days of reperfusion, most infarct microvessels demonstrated significant lectin binding. Our findings suggest that the infarct microvasculature undergoes a transition from an early phase of intense angiogenic activity to a maturation stage associated with pericyte recruitment and formation of a muscular coat. In addition, in the endothelium of infarct microvessels CD31 and CD146 expression appears to precede that of the specific sugar groups that bind the GS-I lectin. Understanding of the mechanisms underlying the formation and remodeling of the microvasculature after MI may be important in designing therapeutic interventions to optimize cardiac repair.
Keywords
A
Effective wound healing is dependent on the formation of a microvascular network capable of supplying the healing infarct with oxygen and nutrients (Tonnesen et al. 2000). Myocardial infarction (MI) is associated with release of angiogenic factors (Li et al. 1996; Lee et al. 2000), which may be critical for neovessel formation in the healing area. In this study we examined the morphological characteristics of the microvasculature in reperfused canine MI. We demonstrated the presence of enlarged pericyte-poor vascular structures in the early stages of healing and noted an increased percentage of pericyte-coated microvessels and a higher arteriolar density during scar maturation. In addition, we found that infarct endothelial cells in the early phase of healing show intense staining for CD31/PECAM-1 and CD146/MEL-CAM but weak Griffonia simplicifolia-I lectin staining, suggesting that PECAM-1 and MEL-CAM expression may precede that of the specific sugar groups that bind the GS-I lectin in the neovessels of healing MI.
Materials and Methods
Ischemia/reperfusion Protocols
Healthy mongrel dogs (15–25 kg) of either sex were surgically instrumented as previously described (Frangogiannis et al. 1998a; 2000a). Anesthesia was induced IV with 10 mg/kg methoexital sodium (Brevital; Eli Lilly, Indianapolis, IN) and maintained with the inhalation anesthetic isoflurane (Anaquest; Madison, WI). A midline thoracotomy provided access to the heart and mediastinum. Subsequently, a hydraulically activated occluding device and a Doppler flow probe were secured around the circumflex coronary artery just proximal or just distal to the first branch. Choice of location depended on the proximity and anatomic arrangement. Indwelling catheters placed in the right atrium, left atrium, and femoral artery allowed blood sampling and pressure monitoring as needed. After surgery, the animals were allowed to recover for 72 hr before occlusion. Coronary artery occlusion was achieved by inflating the coronary cuff occluder until mean flow in the coronary vessel was zero, as determined by the Doppler flow probe. At the end of 1 hr, the cuff was deflated and the myocardium was reperfused. Reperfusion intervals ranged from 7 to 28 days. Circumflex blood flow arterial blood pressure, heart rate, and ECG (standard limb II) were recorded continuously. Analgesia was accomplished with IV pentazocine (Talwin; Winthrop Pharmaceuticals, New York, NY) 0.1–0.2 mg/kg. After the reperfusion periods, hearts were stopped by rapid IV infusion of 30 mEq of KCl and removed from the chest for sectioning from apex to base into four transverse rings ~1 cm in thickness. The posterior papillary muscle and the posterior free wall were identified. Tissue samples were isolated from infarcted or normally perfused myocardium based on visual inspection. Myocardial segments were fixed in B∗5 fixative (Beckstead 1994) for histological analysis. Duplicate adjacent samples were also processed for blood flow determinations using radiolabeled microspheres as previously described (Frangogiannis et al. 1998b). Animals included in the study underwent 7 days (n = 4), 14 days (n = 3), or 28 days of reperfusion (n = 3) and demonstrated evidence of MI based on light microscopic examination of hematoxylin-eosin-stained tissue for replacement fibrosis. Samples described as ischemic were all from areas where ischemic blood flow was less than 25%. Samples of control tissues were taken from the anterior septum and had normal blood flow during coronary occlusion.
Immunohistochemistry
For histological study of cardiac tissue, sections taken from endocardium to epicardium were fixed in B∗5 fixative and embedded in paraffin. Sequential 3–5 μm sections were cut by microtomy. Immunostaining was performed using the ELITE mouse kit (Vector Laboratories; Burlingame CA) as previously described (Frangogiannis et al. 1999, 2000b). Briefly, sections were pretreated with 3% hydrogen peroxide to inhibit endogenous peroxidase activity and incubated with 2% horse serum to block nonspecific protein binding. Subsequently, they were incubated with the primary antibody for 2 hours at room temperature. After rinsing with PBS, the slides were incubated for 30 min with the secondary antibody. The slides were rinsed with PBS and incubated for 30 min in ABC reagent (Hsu et al. 1981). Peroxidase activity was detected using diaminobenzidine (DAB) with nickel (Vector). Slides were counterstained with eosin.
The following primary monoclonal antibodies were used for immunohistochemistry: anti-CD31 monoclonal antibody (Frangogiannis et al. 2000c) (Dako; Carpinteria CA) to label endothelial cells, anti-α-smooth muscle actin antibody (Frangogiannis et al. 1998c) (Sigma; St Louis MO), and the anti-CD146 monoclonal antibody (St Croix et al. 2000) (Chemicon; Temecula CA). Sections incubated with nonimmune serum were used as negative controls. Dual immunohistochemistry was performed using peroxidase-based staining for CD31 followed by alkaline phosphatase-based staining for α-smooth muscle actin developed with the alkaline phosphatase substrate kit I (Vector). Dual stained slides were coverslipped without counterstaining.
Lectin Histochemistry
Lectin histochemistry was performed to identify endothelial cells using the Griffonia simplicifolia lectin I (GS-I) as previously described (Orgad et al. 1984; Alroy et al. 1987). Briefly, sections were pretreated with 3% hydrogen peroxide to inhibit endogenous peroxidase activity and incubated with 2% horse serum. Subsequently they were incubated with biotinylated Griffonia (Bandeiraea) simplicifolia lectin I (Vector) (20 μg/ml with 10 mM HEPES, pH 7.5, 0.15 M NaCl) for 30 min at room temperature. After rinsing with PBS the slides were incubated for 30 min with ABC reagent. Peroxidase activity was detected using DAB with nickel. Slides were counterstained with eosin.
Quantitative Analysis and Statistics
At least two ischemic and two control samples were studied from each experiment. Sections stained using dual immunohistochemistry combining peroxidase-based labeling for CD31 with alkaline phosphatase-based staining for α-smooth muscle actin were photographed with a Leaf MicroLumina digital camera (resolution 300 dpi) mounted on a Zeiss Axioskop microscope. Ten different fields from each control and ischemic sample were analyzed. The percentage of microvessels coated with an α-smooth muscle actin-positive pericyte coat was calculated in the healing infarcts after 7, 14, and 28 days of reperfusion. In addition, the number of arterioles (microvessels fully coated with a muscular coat and with a minor axis >15 μm) was counted in control and fibrotic areas, and expressed as arterioles/mm2 (arteriolar density). Statistical analysis was performed using ANOVA followed by t-test corrected for multiple comparisons (Student-Newman-Keuls). Significance was set at p<0.05.
Results
Morphological Characteristics of Infarct Microvessels
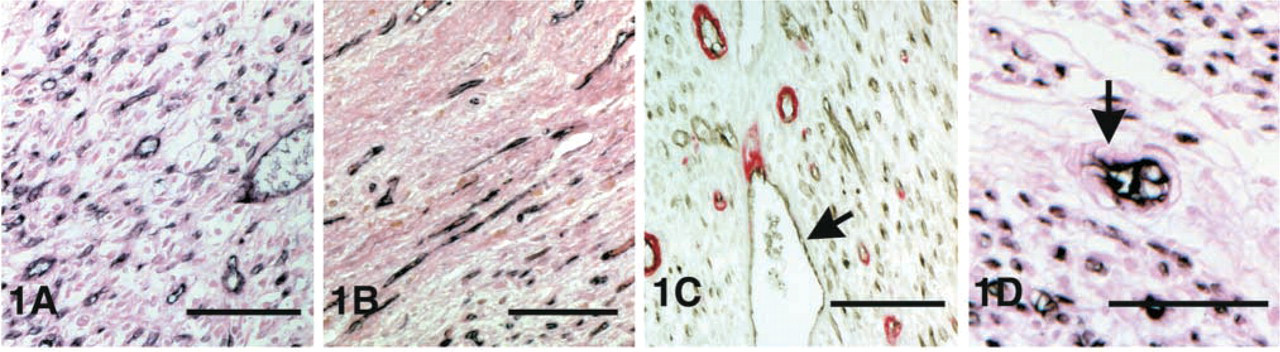
Immunohistochemical staining for CD31 identified the vascular endothelium in control canine myocardium and in the healing MIs. The infarcted territory was highly vascular after 7–14 days of reperfusion (Figure 1A). Infarct microvessels were predominantly capillaries, however the presence of enlarged, pericyte-poor “mother vessels” (Paku and Paweletz 1991; Pettersson et al. 2000) was noted (Figure 1C). In addition, endothelial bridges (Figure 1D) dividing the vascular lumen into multiple smaller channels were occasionally seen. After 28 days of reperfusion the scar contained regions of low vascularity (Figure 1B). This is consistent with our previous findings showing decreasing microvascular density with scar maturation (Frangogiannis et al. 2000c).
Pericyte Coating During Maturation of Myocardial Scars
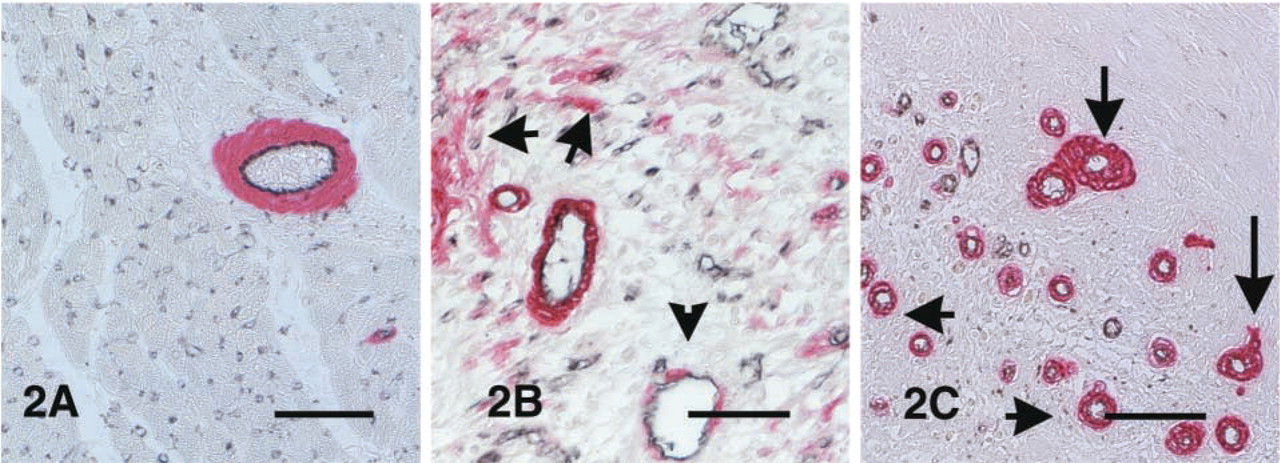
Staining for α-smooth muscle actin identified myofibroblasts and pericytes in the control myocardium and in the healing infarcts. In control areas, α-smooth muscle actin staining was almost exclusively localized in the arteriolar media (Figure 2A). Using dual immunohistochemical staining, we found that in the control canine myocardium less than 1% of the capillaries had an α-smooth muscle actin-positive pericyte coat (Figure 2A).
(
(
(
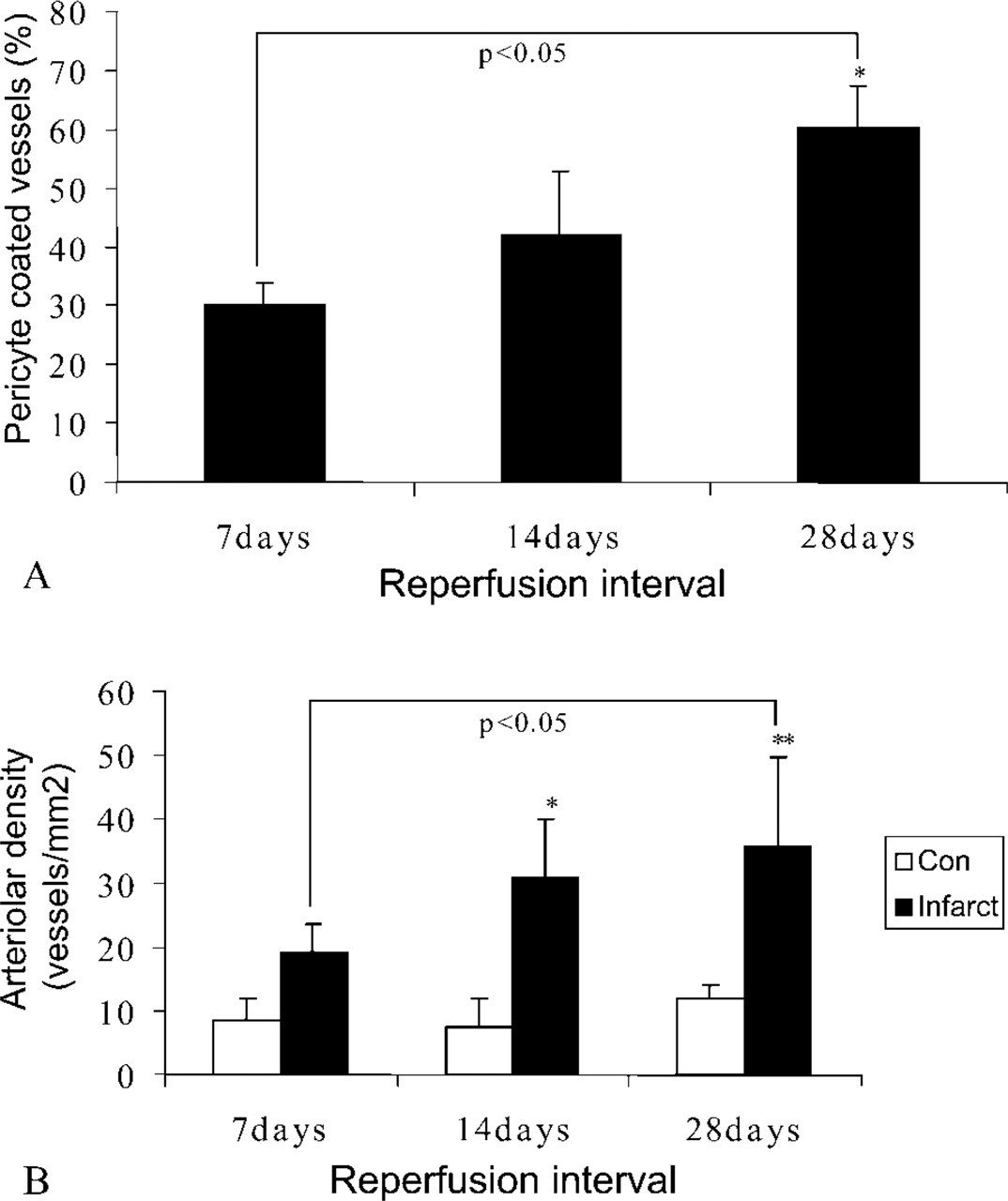
Healing infarcts showed α-smooth muscle actin immunoreactivity in myofibroblast-like cells and perivascular cells (Figure 2B) as previously described (Willems et al. 1994; Frangogiannis et al. 2000c). The wound maturation process was associated with a reduced number of myofibroblasts and a higher percentage of microvessels coated with α-smooth muscle ac-tin-positive pericytes (30.17 ± 3.65% in infarcts after 7 days of reperfusion vs 60.4 ± 6.93% in infarcts after 1 hr of ischemia and 28 days of reperfusion; p<0.05) (Figures 2C and 3A). As the percentage of uncoated microvessels decreased, a significant increase in arteriolar density was noted (35.725 ± 14.05 arterioles/mm2 in infarcts from the 28-day reperfusion group vs 19.11 ± 4.62 arterioles/mm2 in infarcts from the 7-day reperfusion group; p<0.05) (Figure 3B).
Infarct Neovessels Express CD31 and CD146 but Show Weak Staining with GS-I Histochemistry
Serial sections from the canine myocardium were stained for CD31 (PECAM-1), CD146 (Mel-CAM/MUC18), and the GS-I lectin (Figure 4). All three methods were effective in identifying microvessels in control canine myocardium. However, lectin histochemistry gave stronger capillary staining (Figure 4B). In contrast, CD31 staining was much more intense in cardiac arterioles and venules, whereas capillaries showed weaker labeling (Figure 4A). CD146 immunoreactivity was found in endothelial and vascular smooth muscle cells as previously described (Figure 4C). After 1 hr of ischemia and 7 days of reperfusion, the infarct microvessels demonstrated intense staining for CD31 (Figures 5A and 5B) but weak binding with the GS-I lectin (Figures 5C and 5D). CD31 staining was more intense in microvessels of the healing infarct than in control myocardium from the same sections, and this appeared to be the most effective technique in identifying arterioles, venules, and capillaries in the infarcted areas. CD146 immunoreactivity was noted in many microvascular endothelial cells and pericytes of the healing infarct (Figures 5E and 5F). In contrast, after 28 days of reperfusion most of the microvessels in the mature scar showed significant staining for the lectin (Figure 6). These findings suggest that, in MI neovessels, PECAM-1 expression may precede that of the specific sugar groups that bind the GS-I lectin.
Discussion
Wound healing is associated with an angiogenic response, which is critical for scar formation. An increase in hypoxia-inducible factor-1 (HIF-1) is an early response to myocardial ischemia or infarction (Lee et al. 2000) and is followed by induction of VEGF (Dvorak et al. 1995; Cao et al. 1998; Veikkola and Alitalo 1999) in the ischemic areas (Forsythe et al. 1996; Li et al. 1996). In the first few hours after MI, induction of angiostatic factors such as the CXC chemokine interferon γ-inducible protein (IP)-10 is noted and may inhibit neovessel formation until the wound is debrided and a provisional fibrin-based matrix is formed (Frangogiannis et al. 2001). Suppression of IP-10 expression after the first 24 hr of reperfusion presumably allows the active angiogenic phase, during which endothelial cells proliferate (Frangogiannis et al. 1998c) and endothelial progenitor cells infiltrate the healing area (Asahara et al. 1999). We have previously described the dynamic changes in microvessel density during scar formation in reperfused MIs (Frangogiannis et al. 2000c). In addition, Arras and co-workers (1998) have reported, using a porcine model of cardiac microembolization, significant capillary growth during the first 7 days, which decreased after 4 weeks. In this study we examined the structural characteristics of the microvasculature in healing MIs. We found a progressive increase in the percentage of pericyte-coated microvessels and a rise in arteriolar density during cardiac repair. Acquisition of a muscular coat may enhance stability of the vasculature, marking the end of a phase of active angiogenesis and vascular remodeling. We also report that microvessels in canine infarcts showed weak GS-I lectin staining in the early phases of healing, illustrating the phenotypic differences of the vascular endothelium in the healing area. These vessels eventually develop GS-I staining at the late stage of scar formation.
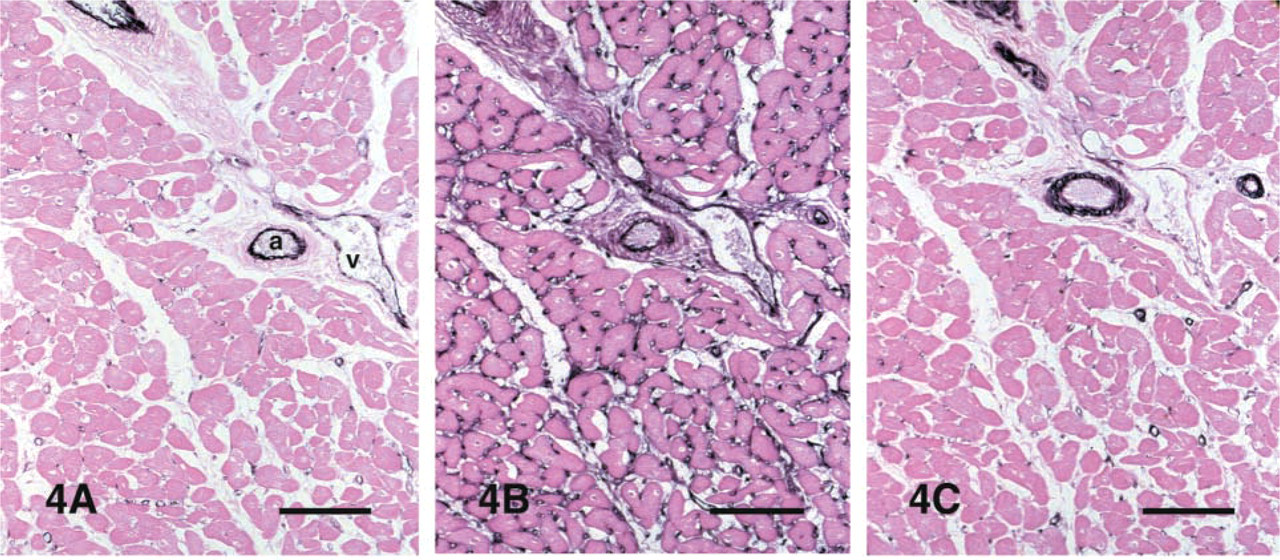
Serial section staining of a control myocardial area for CD31 (
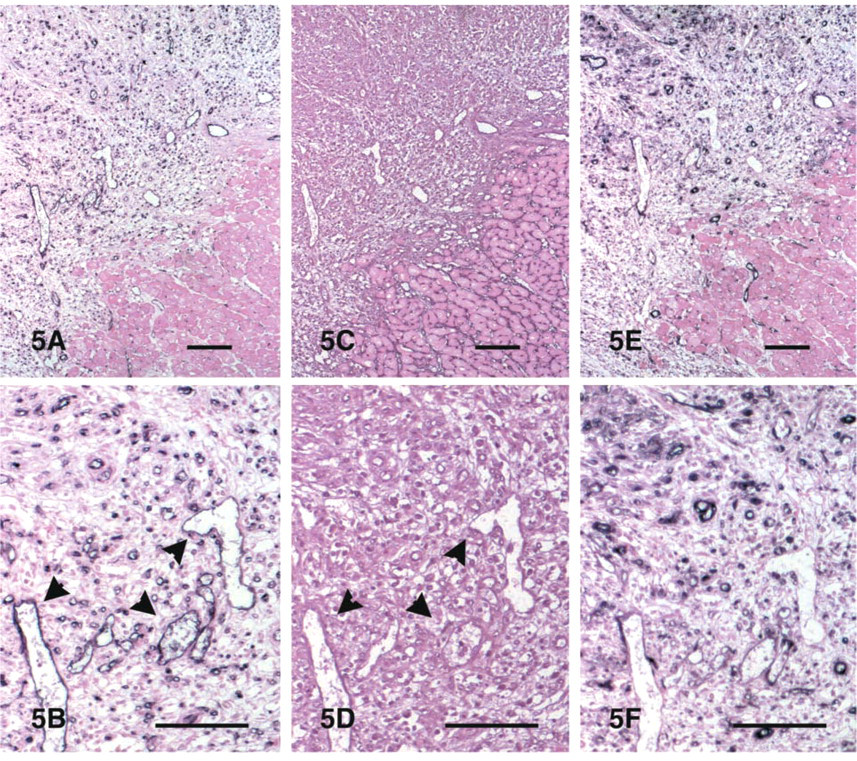
Staining of serial sections of an infarcted area after 1 hr of ischemia and 7 days of reperfusion for CD31 (
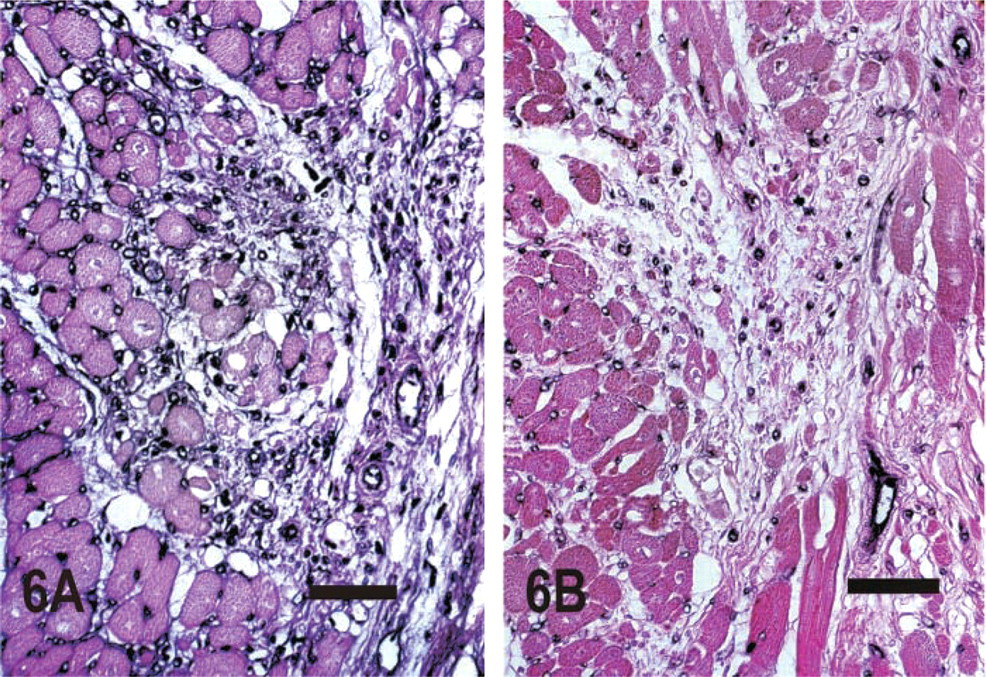
Staining of sections from a canine infarct after 1 hr of ischemia and 28 days of reperfusion with GS-I lectin (
Structural Properties of the Infarct Microvasculature
Angiogenic microvessels demonstrate significant differences in their architecture compared with normal quiescent vessels. Paku and Paweletz (1991) identified enlarged, dilated “mother vessels” with a fragmented basal lamina in tumor-related angiogenesis. Recently, Pettersson and co-workers (2000) described the properties of the new blood vessels induced by VEGF over-expression in normal rat tissues. They reported the transient formation of enlarged, thin-walled pericyte-poor structures, which later evolved into smaller daughter vessels, disorganized glomeruloid bodies, and medium-sized muscular arteries and veins (Pettersson et al. 2000). Generation of dilated enlarged vessels appears to be a characteristic feature of VEGF-mediated angiogenesis in a variety of models (Rosen-stein et al. 1998; Drake and Little 1999; Pettersson et al. 2000).
Healing MIs contain a large number of capillaries in the early stages of healing (Frangogiannis et al. 2000c). Here we describe enlarged pericyte-poor vessels, resembling the “mother vessels” previously described in other models of angiogenesis. Local expression of VEGF may have an important role in the vascular expansion necessary to form these vascular structures. In addition, the relative absence of pericytes is permissive for the angiogenic process and may provide the microvasculature of the infarct with a plasticity window (Benjamin et al. 1998), necessary in this dynamic phase of healing to accommodate the rapidly changing cell populations and metabolic needs. We also noted the occasional presence of transluminal endothelial projections, resembling bridges dividing the vascular structures into multiple lumens (Figure 1C). Scar maturation was associated with the increasing presence of pericyte-coated vessels (Figures 2B and 3) in the infarcted region. In the early stages of healing (7 days of reperfusion), significant numbers of microvessels without an α-smooth muscle actin-positive pericyte coat were noted. In contrast, maturation of the healing infarct was associated with decreased capillarity (Frangogiannis et al. 2000c) (Figures 1B and 2C), an increased percentage of pericyte-coated microvessels (Figure 3A), and a higher arteriolar density (Figure 3B). Investment of growing microvessels with pericytes appears to be coincident with deposition of basement membrane and the cessation of vessel growth (Crocker et al. 1970). Acquisition of a muscular coat may have an inhibitory or suppressive effect on vessel growth. Endothelial cells and pericytes appear to communicate via the release of growth factors that act in a paracrine fashion to influence vascular cell growth and behavior (Hirschi and D'Amore 1996, 1997; Hirschi et al. 1999).
The dynamic structural changes in the infarct microvasculature may have a significant impact on the repair mechanisms after MI. Mother vessels may evolve into muscular arteries and veins, improving blood supply to the injured myocardium, allowing more effective healing, and decreasing infarct extension and ventricular remodeling. Understanding the mechanisms responsible for the formation and maturation of these vascular structures may lead to effective therapeutic strategies aimed at optimizing cardiac repair.
Endothelial Cells in the Early Healing Phase Express CD31 and CD146 but Do Not Show GS-I Lectin Staining
Lectins are glycoproteins, derived from either plant or animal sources, that bind specific carbohydrate moieties of cell surface glycoproteins (Lis and Sharon 1986). Lectin binding intensities may reflect phenotypic changes in the activated angiogenic endothelium of the infarct. Migrating endothelial cells are distinctly hyperglycosylated and express migration-associated glycoproteins (Augustin-Voss and Pauli 1992). Lectin binding was found to be upregulated in the microvascular endothelial cells of the growing corpus luteum (Augustin et al. 1995). However, in a model of rat cornea freeze injury, wound endothelial cells demonstrated decreased binding with wheat germ agglutinin (WGA) and concanavalinA (ConA) (Gordon and Marchand 1990). Furthermore, in the developing rat heart, microvascular CD31 staining preceded GS-I lectin binding, suggesting that PECAM-1 immunoreactivity may be a useful early marker for endothelial cells (Rongish et al. 1995). Our experiments demonstrated intense staining for CD31 in the microvascular endothelium of reperfused canine MIs, which was much more prominent in infarct neovessels than in the vasculature of the control areas. This may reflect the potential role of CD31/PECAM-1 in vessel growth (DeLisser et al. 1997) and tube formation (Yang et al. 1999). In contrast, GS-I lectin staining was intense in the microvascular endothelium of control myocardial areas but not in infarct microvessels after 7 days of reperfusion. Significant lectin binding was noted in more mature scars after 28 days of reperfusion. These findings suggest that changes in lectin binding may reflect the phenotypic modulation of the infarct microvasculature during wound repair.
Our studies also examined the use of CD146 staining to identify the microvasculature in the canine heart. CD146 is a transmembrane glycoprotein constitutively expressed on endothelial cells (Bardin et al. 1996; Shih 1999), with a potential role in cell adhesion and in the rearrangement of the actin cytoskeleton (Anfosso et al. 2001). Infarct microvessels showed CD146 immunoreactivity in all stages of healing.
Conclusions
Vessel growth and maturation are critical for scar formation and cardiac repair. This process represents an important therapeutic target in our efforts to improve post-infarction cardiac recovery (Heymans et al. 1999; Carmeliet 2000). Infarct microvessels undergo phenotypic changes and, after an initial phase of plasticity associated with intense angiogenic activity to meet the metabolic needs of the cellular scar, pericyte recruitment may mark the creation of a more mature vasculature. Understanding the molecular steps involved in this transition may help us design specific interventions to optimize healing of the infarcted myocardium.
Footnotes
Acknowledgements
Supported by NIH Grant HL-42550, the DeBakey Heart Center, and a grant from the Methodist Hospital Foundation (NGF).
We wish to thank Peggy Jackson, Alida Evans, Alejandro Tumang, Stephanie Butcher, and Kathryn Masterman for outstanding technical assistance and Sharon Malinowski and Connie Mata for editorial assistance with the manuscript.