
Abstract
We identified 220-kD protein in bovine skeletal muscle homogenate by affinity chromatography on an agarose column and subsequent SDS-PAGE. Peptide mass fingerprinting (MALDI mass spectrometry) and internal sequence analysis revealed that this protein has homology with several members of the myosin superfamily, particularly with human cardiac β-myosin heavy chain (β-MHC). A rabbit polyclonal antibody against the 220-kD protein specifically stained a 220-kD band in Western blots of skeletal muscle homogenate. Immunohistochemical experiments on cryostat sections demonstrated that in skeletal muscle this protein is exclusively localized at the neuromuscular junctions, no immunoreactivity being present at the myofibril level. Because of its relative homology with cardiac β-MHC, we also investigated the distribution of the 220-kD protein in bovine heart. In cardiac fibers, 220-kD protein-related immunoreactivity was restricted to the intercalated disks, whereas myofibrils were completely devoid of specific immunoreactivity. This distribution pattern was completely different from that of cardiac β-MHC, which involved myofibrils. Because of the above biochemical and immunohistochemical features, the 220-kD protein we have identified is suggested to be a novel member of the non-muscle (non-sarcomeric) myosin family.
M
Although the precise function of the non-sarcomeric myosins (generally called non-muscle myosins) remains to be determined, they appear to play a role in specialized cell functions such as membrane trafficking, cell movements, and signal transduction (Mermall et al. 1998; Lionne et al. 2001). However, despite the many studies reported in the literature, only three non-muscle myosins have been well characterized to date. Among these, in vertebrates, most tissues contain two different isoforms: non-muscle myosin heavy chain (NMHC) II-A and II-B (Sellers 2000; Straussman et al. 2001). The genes for these proteins are expressed in a tissue-specific (Katsuragawa et al. 1989; Kawamoto and Adelstein 1991; Murakami et al. 1991; Phillips et al. 1995) and differentiation-dependent manner (Murakami et al. 1993; Itoh and Adelstein 1995; Kawamoto 1996; Takahashi et al. 1999). NMHC II-A is predominant in platelets (Maupin et al. 1994; Pecci et al. 2002), fibroblasts (Saitoh et al. 2001), endothelial cells (Kolega 1998), and in leukocytes and smooth muscle (Choi et al. 1996; Pecci et al. 2002). NMHC II-B is mainly present in cardiac myocytes (Tullio et al. 1997; Uren et al. 2000) and brain (Tullio et al. 2001). Although there is some overlap in their intracellular distribution, NMHC II-B is localized near the plasma membrane of some cells (Xenopus A6, HeLa, and melanoma cells), whereas NMHC II-A is found in stress fibers. This suggests that each isoform might play different functions (Maupin et al. 1994; Rochlin et al. 1995; Kelley et al. 1996; Kolega 1998; Wylie et al. 1998).
The third non-muscle myosin that has been extensively characterized is myosin-Va (Reck-Peterson et al. 2000). This protein has been first cloned and sequenced from a chicken brain library (Espreafico et al. 1992). Recent evidence suggests that this unconventional myosin may play a role in organelle transport or membrane targeting (Cheney and Rodriguez 2001; Karcher et al. 2002; Schott et al. 2002). The expression of myosin Va is high in cell bodies and dendrites of Purkinje cells. In primary cultures of glia and neurons, myosin Va has a more abundant distribution in the region of the Golgi complex and at the tips of long processes, such as the growth cones (Espreafico et al. 1992; Suter et al. 2000). Interestingly, myosin Va is also an abundant component of the postsynaptic density of CNS synapses (Walikonis et al. 2000).
Here we discuss the identification of a 220-kD protein at the neuromuscular junction, showing biochemical and immunohistochemical features of a novel non-muscle myosin heavy chain (NMHC).
Materials and Methods
Protein Identification
Specimens of bovine skeletal muscle were taken from adult animals at a local slaughterhouse. Samples were immediately snap-frozen in liquid nitrogen and stored at −80C until used. In a typical experiment, approximately 80–100 g of muscle was homogenized in 50 mM Tris, pH 7.5, containing 0.15 M NaCl (1:5 w:v), using an UltraTurrax T25 homogenizer (IKA; Staufen, Germany). The homogenate was ultracentrifuged at 105,000 X g for 1 hr at 4C and soluble proteins were partially precipitated with 50% ammonium sulfate. The resulting pellet was resuspended in 50 mM Tris (pH 7.5) containing 0.15 M NaCl and 0.005% Brij-35, and was applied at 4C to a small column (2 ml) of agarose (Sepharose 4B; Pharmacia Biotech, Uppsala, Sweden). After sample application, the column was washed (10 ml/hr) with 10 volumes of equilibrium buffer and finally eluted with 0.05% trifluoroacetic acid (TFA), pH 2.5, containing 0.15 M NaCl. To neutralize the acid pH of the eluent, the eluted material (0.5 ml/tube) was immediately mixed with aliquots of 1 M Tris, pH 7.5 (0.04 ml/tube).
Protein concentration was determined by the method of Bradford (1976), using a commercial kit (Bio-Rad Laboratories; Hercules, CA) with BSA as a standard. Conventional 1% SDS-PAGE was performed on 12.5% precast gels (ExcelGel; Pharmacia Biotech) using a horizontal apparatus (LKB Multiphor II electrophoresis system (Pharmacia Biotech). Gels were stained with Coomassie Blue (R250; Farmitalia Carlo, Erba, Italy) or by using a silver staining kit (Pharmacia Biotech). Standards for estimation of molecular weight were purchased from Pharmacia Biotech. Densitometry of electrophoresed bands was performed using a PC-assisted CCD camera (gel Doc 2000 system/Quantity One software; BioRad Laboratories).
Mass Spectrometry and Sequence Analysis
The mixture of proteins eluted from the Sepharose 4B column was electrophoresed on a 7% polyacrylamide gel containing 1% SDS by using a vertical apparatus (Hoefer Pharmacia Biotech; San Francisco, CA) (1.5 mm gel thickness). The gel was stained with Coomassie Blue R250 for 10 min to visualize the bands and then was de-stained by soaking for 3 hr to remove the excess stain. The band of interest (220 kD) was excised from the gel and subjected to trypsin digestion according to Williams et al. (1996). As a control, an identical section of the same gel that did not contain any protein was also digested.
A small aliquot of the gel digested with trypsin was subjected to matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) (Keck Facility; Yale University, New Haven, CT). MALDI-MS was carried out on a Micromass TofSpec SE mass spectrometer equipped with delayed extraction and a reflection. To attain the high level of accuracy needed for peptide mass searching, 100 fml bradykinin with a protonated, monoisotopic mass of 1060.57, and an ACTH clip, with a protonated, monoisotopic mass of 2465.2, were added as external calibrants.
The resulting peptide masses were then subjected to peptide mass identification using PeptIdent (at the EBI) and Pro-Found (at Rockefeller University). Peptide masses were compared with the theoretical masses derived from the sequence contained in SWISS-PROT/TrEMBL databases (release 40.16 and 20.4). The search parameters used were as follows: cysteines unmodified, maximum allowed peptide error 0.1 Dalton.
For amino acid sequence, the tryptic mixture was separated using a Labservice Analytica chromatographer (model LabFlow 4000) on a C18 column (250 mm X 2.1 mm; 5-μm Vydac) in a linear gradient of acetonitrile (1–70%) in 0.05% TFA. Amino acid sequence was carried out using a sequencer equipped with an on-line HPLC system (Keck Facility, Yale University).
Antibody Preparation
For antigen preparation, the protein mixture eluted from the Sepharose 4B column was subjected to semipreparative SDS-PAGE on 12.5% gels by using a vertical apparatus (Hoefer Pharmacia Biotech; San Francisco, CA) (1.5 mm gel thickness). After electrophoretic separation, the gels were stained faintly with Coomassie Blue and the 220-kD band was excised and used to immunize two New Zealand White rabbits. For each injection two or three slices (containing approximately 200 μg antigen) were pooled, homogenized in 10 mM Tris, pH 7.5 (1 ml), and emulsioned with an equivalent amount of incomplete Freund's adjuvant (Sigma; St Louis, MO). For the first injection, complete Freund's adjuvant (Sigma) was used. Samples were injected into a rabbit's back by multiple intradermal injections (20–30 injections). Four antigen administrations were performed at 28–30-day intervals.
Western Blotting
Immunoblotting experiments were performed essentially according to Towbin et al. (1979). In particular, SDS-PAGE was performed as described above using a LKB Multiphor II electrophoresis system (Pharmacia Biotech). Samples of skeletal muscle homogenate, corresponding to 10–15 μg protein, were loaded on a single lane. Protein transfer to nitrocellulose paper (Schleicher & Schuell; Dassel, Germany) was obtained by a semidry system (Novablot; Pharmacia Biotech) using a continuous buffer (39 mM glycine, 48 mM Tris) containing SDS (0.037% w/v) and methanol (20% v/v). To reduce the background, the blotted membranes were treated with 5% skim milk, immersed for 20 min in an avidin blocking solution (Vector; Burlingame, CA), washed for 5 min with TBS, and finally incubated (20 min) in a biotin blocking solution (Vector). The membranes were then incubated (1 hr at RT) with anti-220-kD protein immunoglobulins (Igs) purified from the rabbit antisera by ion-exchange chromatography on a DEAE-Affigel blue column (Bio-Rad Laboratories). Concentrations of 1–3 μg/ml of specific Igs were used. Controls were done substituting specific Igs with equivalent amounts of nonspecific (pre-immune) Igs. The membranes were then washed with TBS-Tween 0.1% (four 5-min changes) and treated with the secondary biotinylated antibody (1 hr at RT). After a further wash, a final step (30 min) was performed by using an avidin-biotin–peroxidase complex procedure (Vectastain Elite ABC kit; Vector). A peroxidase substrate kit (Vector) containing diaminobenzidine (DAB) and a nickel solution was used to enhance staining sensitivity.
Immunohistochemistry
For immunohistochemical experiments, small blocks of skeletal and cardiac muscle were embedded in gum tragacanth (Merck; Darmstadt, Germany) and snap-frozen in isopentane precooled with liquid nitrogen. Serial sections (8–10 μm thick) were cut in a cryostat at −20C and mounted on gelatin-coated glass slides. Specimens were fixed for 4 min at RT with 4% paraformaldehyde (Merck), quenched with 0.1 M glycine-HCl, pH 7.4, and treated with 3% H2O2 in methanol to inhibit endogenous peroxidase. After extensive washes in PBS, sections were preincubated for 1 hr with 6% nonfat dry milk and incubated with the appropriate antibody up to 72 hr at 4C.
Anti-220-kD protein Igs were isolated on a DEAE-Affigel blue column and used at a final concentration of 1–10 μg/ml. A commercial monoclonal antibody to human cardiac MHC, type α/β (Biocytex Biotechnol; Marseille, France) was also used at a final dilution of 1:100. Immunohistochemical distribution was revealed according to a standard avidin-biotin–peroxidase method. Briefly, after washes with PBS–0.3% Triton and PBS alone, slides were incubated (1 hr at RT) with a biotinylated secondary antibody (Vector), washed again, and incubated (30 min at RT) with an avidin-biotin–peroxidase complex (Vectastain Elite ABC kit; Vector). Finally, sections were washed and treated with 0.05% 3–3′ diaminobenzidine and 0.015% H2O2.
Negative controls were done by omission of the first or the secondary antibody or by replacing the primary antibody with an equivalent amount of Igs purified from the preimmune sera. In additional experiments, specific anti-220-kD protein Igs were preabsorbed with saturating amounts of purified antigen (antigen:antibody ratio=5).
Results
Protein Identification and Sequence Analysis
Samples of bovine skeletal muscle were homogenized and precipitated with 50% ammonium sulfate. The resulting pellet was then resuspended in the equilibrium buffer and applied to an agarose (Sepharose 4B) column that is known to selectively bind carbohydrate recognition molecules (Alliegro and Linz 1997). After extensive washes with the equilibrium buffer, the proteins retained on the Sepharose column were eluted with 0.05% TFA (pH 2.5). Recovery approximated 10–20 μg/100 g wet weight of the original tissue. SDS-PAGE (Figure 1) showed that, under our experimental conditions, the protein mixture retained by the column was mainly constituted of two prominent bands of 220 and 45 kD, respectively. Densitometric analysis of the gels stained with R250 Coomassie Blue revealed that the 220-kD and 45-kD bands represented about 40% and 10–20% of the total proteins recovered from the column, respectively.
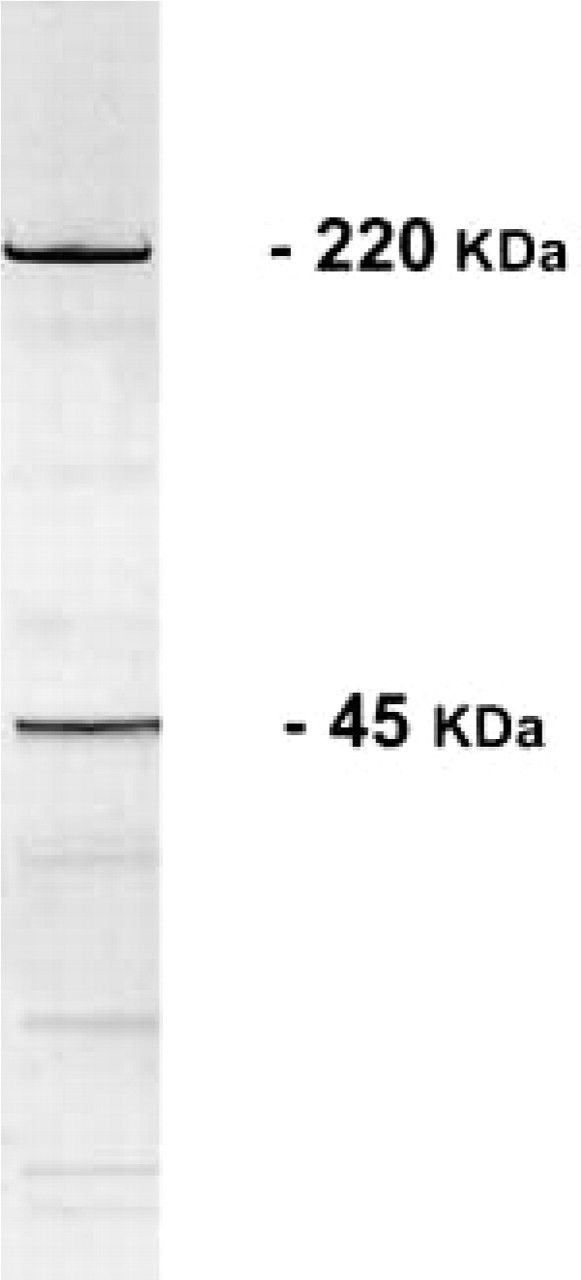
SDS-PAGE of a protein fraction of skeletal muscle homogenate bound to an agarose column. Elution was obtained by 0.05% TFA. Two prominent bands (220 kD and 45 kD) are apparent.
For protein identification, a sample of the 220-kD band was excised and digested with trypsin. MALDI-MS was carried out on approximately 5% of the tryptic digest. MALDI-MS detected a clear break in the number of peptides matched, with a sufficient number of masses for database searching. The primary program we used for searching was PeptIdent, which relies on SWISS-PROT/TrEMBL databases. This search revealed that our protein had a top score to a myosin heavy chain of various sources, mainly cardiac isoforms. Furthermore, automated Edman degradation of a purified tryptic peptide gave the following sequence: STHPHFVR. This sequence is identical to that present in human cardiac β-MHC in position 664–671. This sequence is conserved in all the other cardiac β-myosins from various sources whose complete sequences are known, but is not conserved in the primary structure of skeletal myosins.
Immunochemistry and Immunohistochemistry
To study our protein further, we generated a polyclonal antibody in rabbits using the entire protein as antigen (for details see Materials and Methods). The resulting Igs purified from rabbit sera specifically stained a single band of 220 kD in Western blots of muscle homogenate (Figure 2). Furthermore, by using the same Igs we studied the immunohistochemical distribution of the 220-kD protein on cryostatic sections of skeletal and cardiac muscles. In bovine skeletal muscle anti-220-kD protein Igs stained specifically scattered structures near the skeletal muscle fibers, the sarcoplasm of all muscle fibers being completely devoid of immunoreactivity (Figure 3A). In adjacent sections, a classical cholinesterase staining (Tago et al. 1986) allowed us to identify those positive areas as neuromuscular junctions (Figure 3B). Control experiments (for details see Materials and Methods) were completely negative (Figure 3C).

Immunoblotting analysis of 220-kD protein-related immunoreactivity in skeletal muscle homogenate. (
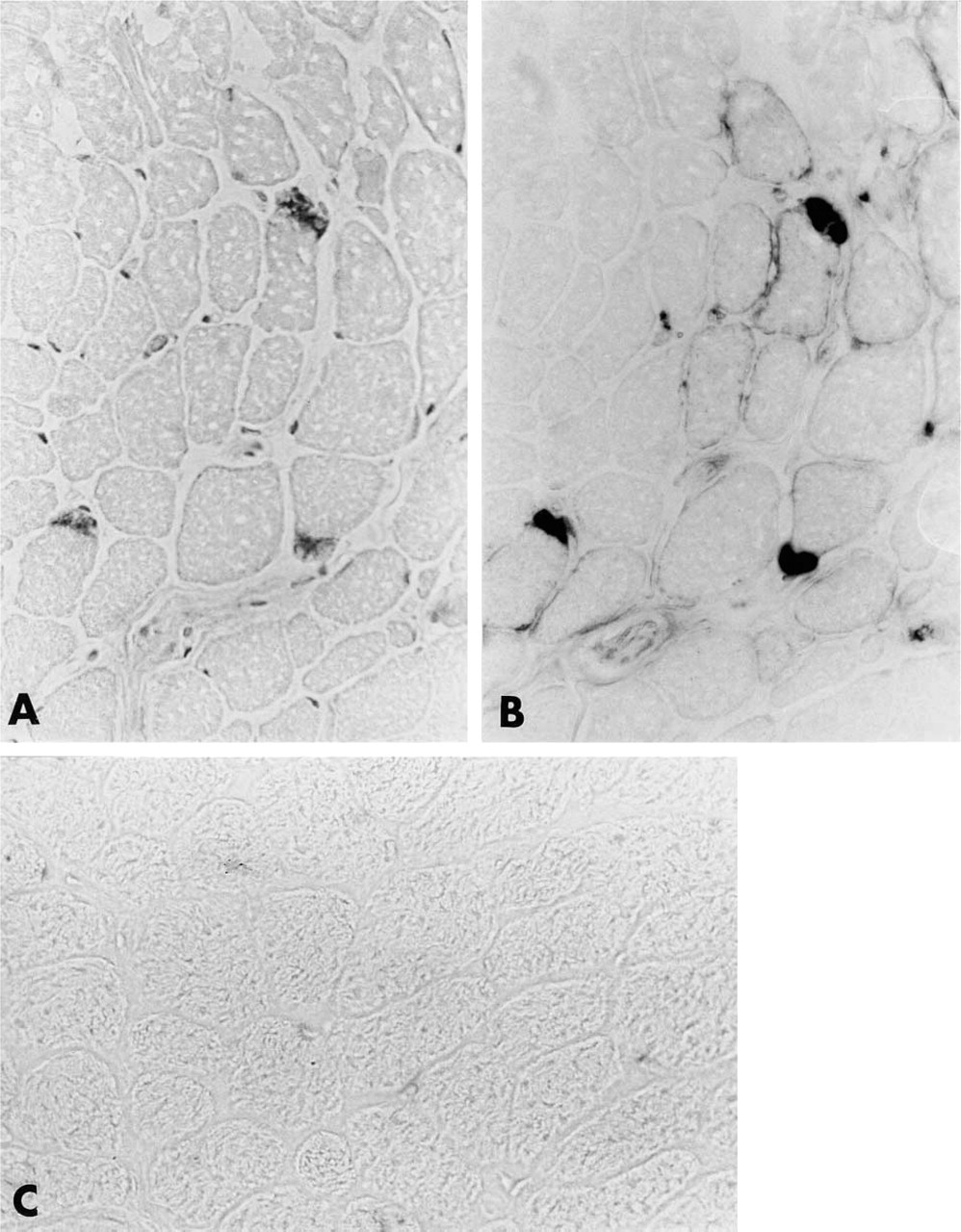
Immunoperoxidase localization of 220-kD protein-related immunoreactivity in bovine skeletal muscle. Specific labeling occurs in small areas adjacent to muscle fibers (
In bovine heart, anti-220-kD protein Igs selectively labeled the intercalated disks of all myocardial cells (Figure 4A). Control experiments were completely negative (Figure 4B). A very different pattern of immunostaining was obtained by using a monoclonal antibody specific for cardiac MHC. In bovine skeletal muscle, anti-cardiac MHC Igs strongly and specifically stained the cytoplasm of only few scattered muscle fibers (Figure 5A). By contrast, as expected, in bovine heart all cardiac fibers were specifically labeled (Figure 5B).
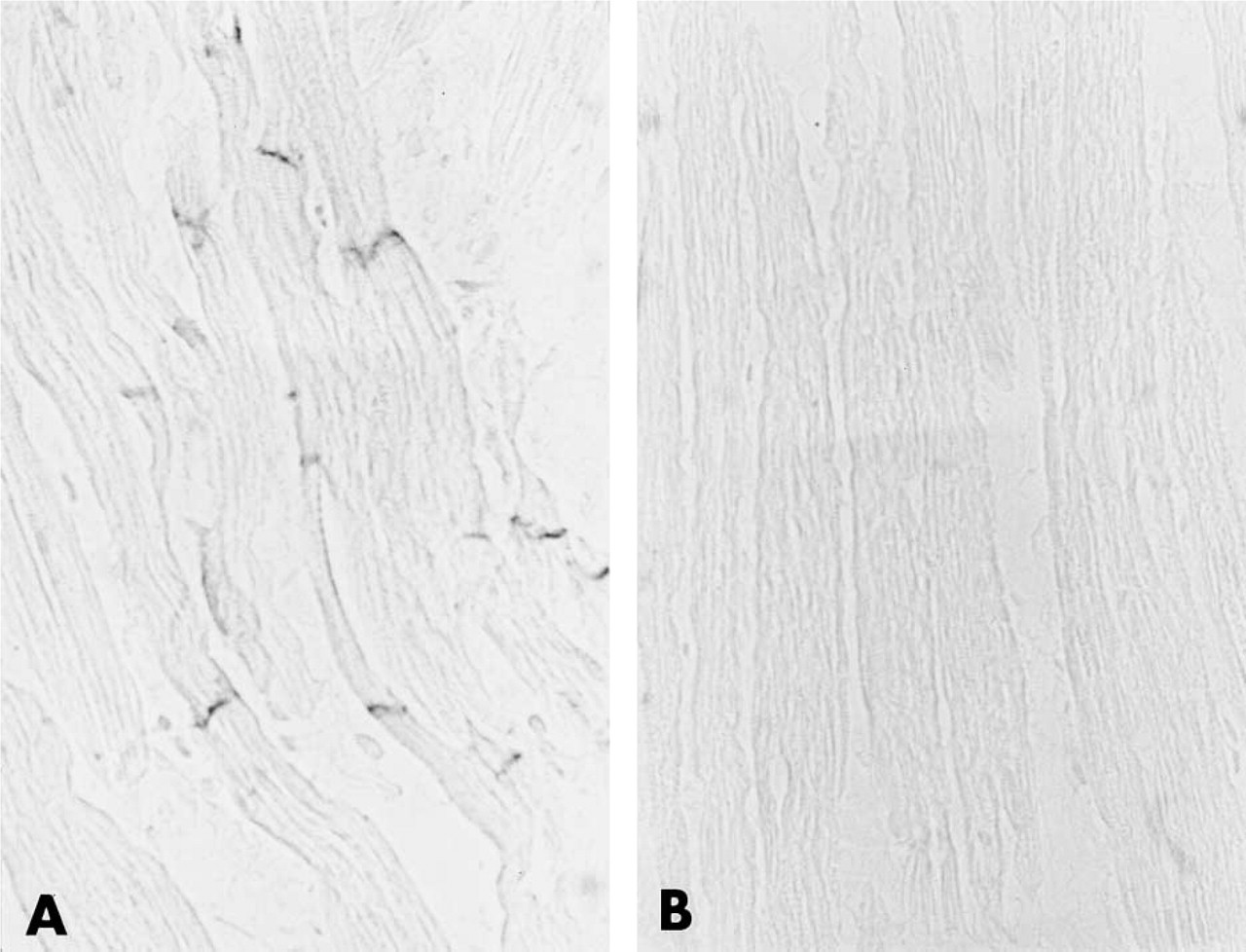
(A) Immunoperoxidase localization of the 220-kD protein-related immunoreactivity in bovine heart. Specific immunoreactivity is present exclusively in the intercalated disks. (
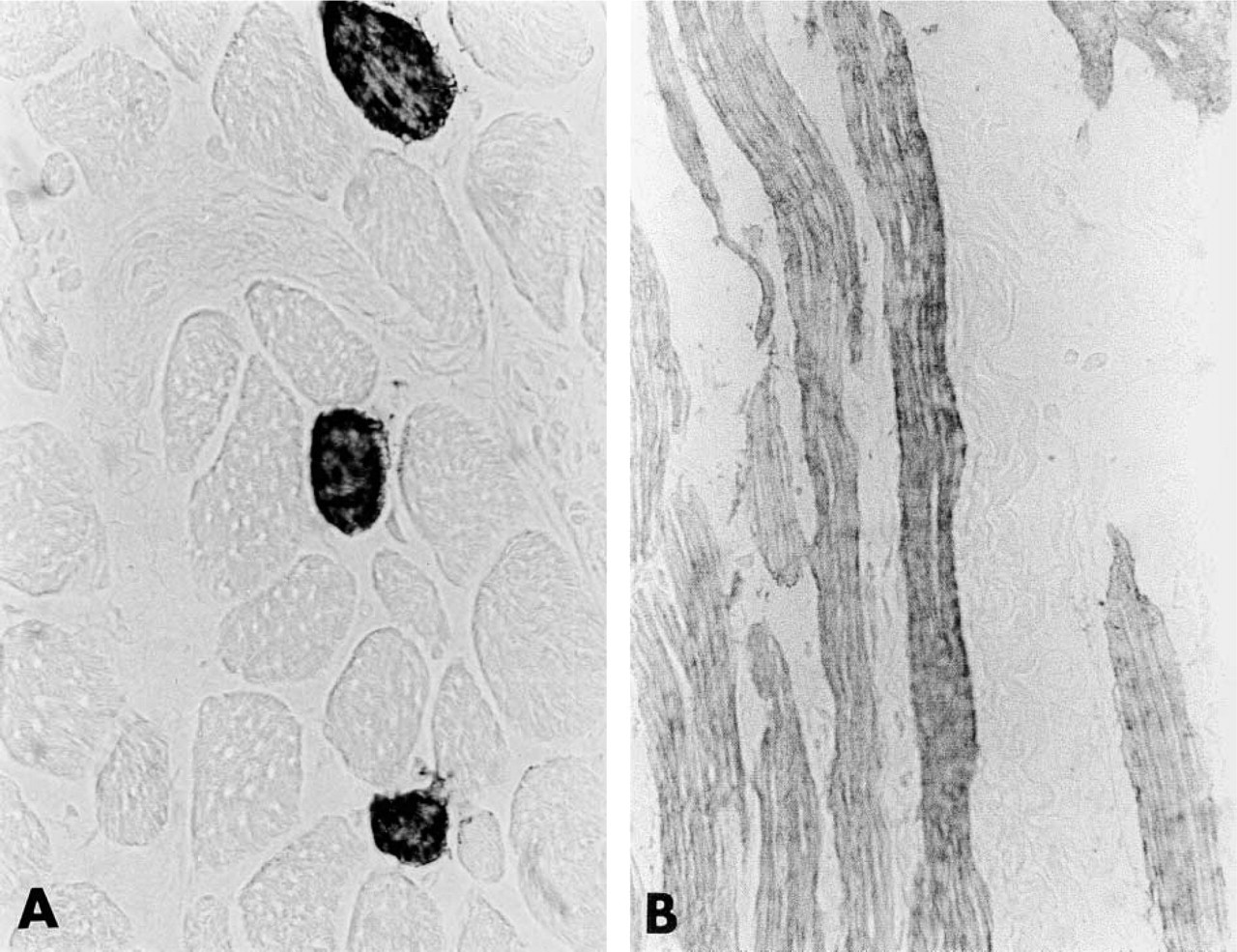
(A) Immunoperoxidase localization of cardiac myosin heavy chain in bovine skeletal muscle. Only a few scattered muscle fibers are labeled. (
Discussion
The first molecular motor to be discovered was conventional, two-headed, and filament forming myosin II. Because of its important role in muscular contraction, myosin II has been extensively studied at both the molecular and the cellular level.
Myosin II has also been detected in non-muscle cells. In particular, vertebrates have two NMHC genes, encoding for NMHC II-A and NMHC II-B (Sellers 2000; Straussman et al. 2001). In the past few years, many novel myosins have been discovered and at least 15 structurally distinct classes have been identified. However, most of these myosins have not been studied in detail, although it is clear that they are involved in a variety of cell processes (Reck-Peterson et al. 2000; Sellers 2000).
Here we have described the identification, the initial characterization, and the immunohistochemical localization of a 220-kD protein that is probably an additional member of this large superfamily. This protein has been isolated from bovine skeletal muscle homogenate by affinity chromatography on an agarose column. This approach has also been used by other workers to purify other NMHCs from mammalian retinal pigment epithelial and endothelial cells (Alliegro and Linz 1997). Like other known MHCs, our protein displays a molecular weight of 220 kD in SDS-PAGE. Furthermore, MALDI-MS analysis and sequence data demonstrated a homology of this protein with well-known members of the myosin family, in particular with human cardiac β-MHC. To further investigate this homology, we performed a series of immunohistochemical experiments in adult bovine skeletal and cardiac muscle using our anti-220-kD protein antibody and an antibody specific for cardiac MHC. In line with literature data, the anti-cardiac MHC antibody gave strong specific immunostaining in cardiac cells, which was mainly associated with myofibrils (Weiss and Leinwand 1996), whereas only scattered fibers in skeletal muscle were positive. This finding is not surprising because some skeletal muscle fibers (type I) are known to express cardiac β-MHC (Weiss and Leinwand 1996). Conversely, a very different distribution pattern occurred for the 220-kD protein, which was localized selectively at the neuromuscular junctions in skeletal muscle and at the intercalated disks in cardiac muscle. These results indicate that our protein is distinct from cardiac β-MHC (which is localized in myofibrils) and is a non-muscle (non-sarcomeric) MHC.
Several studies have reported the presence of NMHCs Va and VI in neurons and synapses of the CNS (Espreafico et al. 1992; Suter et al. 2000). For the neuromuscular junction, to our knowledge our 220-kD protein represents the first example in the literature of an NMHC localized at that level. Because our MALDI-MS and sequence experiments did not show any significant homology of our 220-kD protein with myosin Va and VI, it appears likely that our protein represents an additional member of the “synaptic” NMHC family. However, the function(s) of these “synaptic” myosins remains unknown at present. In this context, however, myosin Va has been postulated (as an organelle motor) to be involved in synaptic transmission and plasticity, but recent experimental evidence did not confirm this hypothesis (Schnell and Nicoll 2001).
In the myocardium, our anti-220-kD protein antibody stains exclusively the intercalated disks, where NMHC II-B has been also detected (Takeda et al. 2000). However, NMHC II-B, is also present in myofibrils, co-localizing with NMHC II-A at Z-lines (Takeda et al. 2000), i.e., the sites that are negative for our protein. These immunohistochemical data exclude the possibility that our antibody crossreacts with NMHC II-A and/or II-B. On the other hand, MALDI-MS and sequence data also do not indicate any significant homology of our protein with NMHC II-A and B. Taken together, the present results reinforce the idea that our 220-kD protein may represent a novel isoform of NMHC.
Footnotes
Acknowledgements
Supported by the Italian Ministry of University and Technology (Grants University La Sapienza, Facoltà and Ateneo to L. Fumagalli).